MEEP was developed in the groups of Steven G Johnson, John D. Joannopoulous, and Marin Soljačić at MIT. Nanostructures & Computation Wiki at MIT.
Meep is dependent upon several libraries and other packages. The easiest way to set up the environment correctly for Meep is to type 'module load meep', which will load the latest serial (single-threaded) version. To see available versions, type 'module avail meep'.
Note: The h5utilsg are often used with Meep. These are only available for v1.4.3 and not the older 1.2.1 version. Therefore, to use the h5utils, you will need to load the 1.4.3 version of Meep as in the example below.
he following sample batch scripts are taken from the Meep tutorials. Create a batch script along the following lines: (Meep control file: straight_waveguide.ctl).
#!/bin/bash
# this file is called meep.bat
module load meep/1.2.1/serial
cd /data/$USER/mydir
meep straight_waveguide.ctl
module load meep/1.4.3/mpi #this will automatically unload the 1.2.1/serial version
h5topng -S3 -Zc dkbluered -a yarg -A \
straight_waveguide-eps-000000.00.h5 straight_waveguide-ez-000200.00.h5
A serial Meep run will only utilize one core. From previous experience, this run will require very little memory, certainly less than 1 GB. Thus, it can be submitted to the default 1 core, 1 GB memory. Submit this job with:
sbatch meep.bat
If your own runs require more memory, submit with something like:
sbatch --mem-per-cpu=#g meep.bat
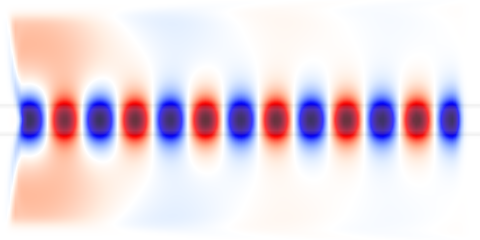
This job produces two .h5 output files and the following png image:
Create a batch script along the following lines: (Meep control file: bent_waveguide.ctl) Use the appropriate module for the network (gige or ib) on which you want to run.
#!/bin/bash # this file is called meep.par.bat module load meep/1.2.1/mpi cd /data/$USER/mydir mpirun `which meep-mpi` bent_waveguide.ctl # h5utils are only available for the later versions, so switch modules module load meep/1.4.3/mpi h5topng -t 0:329 -R -Zc dkbluered -a yarg -A bent_waveguide-eps-000000.h5 bent_waveguide-ez.h5 convert bent_waveguide-ez.t*.png ez.gif
Submit this job with:
sbatch --ntasks=# --constraint=x2650 --exclusive meep.par.batThis will submit the job to # cores. Note that this number does not have to be specified within the batch script, as OpenMPI will get the number of allocated cores from Slurm automatically.
If running on more than 1 node, the parameter ''--constraint=x2650" ensures that the job will run on nodes of the same processor speed. The parameter '--exclusive' ensures that the job will allocate the nodes exclusively. This is recommended for parallel jobs that have inter-node communication.
This job produces two .h5 files, a series of png images, and the animated gif below:

As with all parallel jobs, it is advisable to run benchmarks to determine the appropriate number of MPI processes/cores for a job. For example, you could run the following series of tests with a small job:
sbatch --ntasks=1 --constraint="x2650" --exclusive meep.par.bat sbatch --ntasks=2 -constraint="x2650" --exclusive meep.par.bat sbatch --ntasks=4 -constraint="x2650" --exclusive meep.par.bat sbatch --ntasks=8 -constraint="x2650" --exclusive meep.par.bat sbatch --ntasks=16 -constraint="x2650" --exclusive meep.par.bat
The '--constraint="x2650" parameter' ensures that all the jobs will run on the same processor type. '--exclusive' ensures that no other jobs will run on those nodes, as is best for parallel jobs that have inter-node communication.
Note down the elapsed time (reported in the Meep standard output) for each run, and calculate the efficiency for each run as:
100 * Time on 1 core
Efficiency = ---------------------------
n * Time on n cores
You should then choose the number of cores where the efficiency is at least 760%, to get the best improvement without wasting resources for your production jobs.
An example benchmark using the bent_waveguide control script.
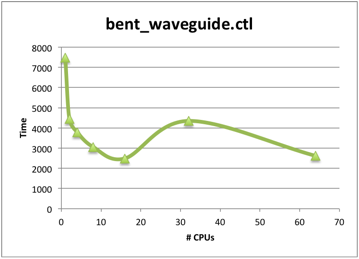
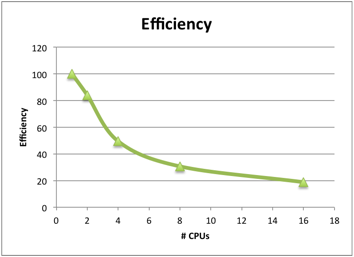
In this example, it would only be appropriate to run on 2 cores, since the efficiency drops below 60% for any larger number of cores. However, other Meep runs are known to scale better.
Swarm' would be appropriate for a group of serial Meep runs.
For a swarm of single-threaded Meep jobs, set up a swarm command file along the following lines:
# this file is swarm.cmd cd /data/$USER/mydir; meep file1.ctl cd /data/$USER/mydir; meep file2.ctl cd /data/$USER/mydir; meep file3.ctl [...etc...]
Submit this swarm of single-threaded jobs with:
swarm -f swarm.cmd --module meep/1.2.1/serial
You can also add a '-g #' switch if each process requires more than 1 GB of memory.
You would probably do this only for testing purposes. Allocate an interactive node, load the module, and run your meep command. Sample session:
[user@biowulf]$ sinteractive
salloc.exe: Granted job allocation 9107
slurm stepprolog here!
[user@p20]$ module load meep/1.2.1/serial
[user@p20]$ meep straight_waveguide.ctl
-----------
Initializing structure...
Working in 2D dimensions.
Computational cell is 16 x 8 x 0 with resolution 10
block, center = (0,0,0)
size (1e+20,1,1e+20)
axes (1,0,0), (0,1,0), (0,0,1)
dielectric constant epsilon diagonal = (12,12,12)
time for set_epsilon = 0.0544579 s
-----------
creating output file "./straight_waveguide-eps-000000.00.h5"...
creating output file "./straight_waveguide-ez-000200.00.h5"...
run 0 finished at t = 200.0 (4000 timesteps)
Elapsed run time = 2.86689 s
[user@p20 meep]$ exit
exit
slurm stepepilog here!
salloc.exe: Relinquishing job allocation 9107
[user@biowulf]$
Meep Manual at the MIT website.