OpenCRAVAT is a new open source, scalable decision support system for variant and gene prioritization. It includses a modular resource catalog to maximize community and developer involvement, and as a result the catalog is being actively developed and growing every month. Resources made available via the store are well-suited for analysis of cancer, as well as Mendelian and complex diseases.
OBAllocate an interactive session and run the program. Sample session:
[user@biowulf]$ sinteractive --mem=16g --gres=gpu:p100:1,lscratch:10 -c4 --tunnel
...
salloc: Granted job allocation 61014005
salloc: Waiting for resource configuration
salloc: Nodes cn0843 are ready for job
Created 1 generic SSH tunnel(s) from this compute node to
biowulf for your use at port numbers defined
in the $PORTn ($PORT1, ...) environment variables.
Please create a SSH tunnel from your workstation to these ports on biowulf.
On Linux/MacOS, open a terminal and run:
ssh -L 40837:localhost:40837 denisovga@biowulf.nih.gov
For Windows instructions, see https://hpc.nih.gov/docs/tunneling
[user@cn2389 ~]$
Store the PORT1 number and the node_id you've got, in this case [user@cn2389 ~]$ module load OpenCRAVAT/2.12.0 [+] Loading annovar 2020-06-08 on cn2389 [+] Loading OpenCRAVAT 2.12.0 ...In order to annotate and interpret variants, OpenCRAVAT (OC) makes use of a database comprising chanks of data called "modules" (not to be confused with the Biowulf modules). The currenly installed modules OC modules are available in the folder: $OC_MODULES.
[user@cn2389 ~]$ cd /data/$USER [user@cn2389 ~]$ mkdir OpenCRAVAT && cd OpenCRAVAT [user@cn2389 ~]$ ln -s /usr/local/apps/OpenCRAVAT/2.12.0/src/cravat/logs [user@cn2389 ~]$ oc new example-input . example_inputcat example_input | wc -lThe latter command will create a file "example_input" in your current directory:
[user@cn2389 ~]$ cat example_input | wc -l 48head -n 20 example_input [user@cn2389 ~]$ head -n 20 example_input chr11 108244076 + C G s0 chr17 31350290 + C T s0 chr7 55174772 + GGAATTAAGAGAAGC - s0 chr7 140753336 + AC TT s0 chr3 179234296 + C T s0 chr4 54733174 + T A s0 chr9 21970902 + C A s0 chr3 10149836 + G C s0 chr3 179199156 + A G s0 chr17 7674953 + T A s0 chr5 112839543 + G C s0 chr7 55174015 + G C s0 chr21 34880581 + T C s1 chr17 7674241 + G A s1 chr3 41224571 + C T s1 chr3 10149886 + T A s1 chr11 534286 + C A s1 chr17 7674237 + G C s1 chr3 41224610 + C A s1 chr17 7675214 + A T s1Run OpenCRAVAT on the test input file:
[user@cn2389 ~]$ oc run ./example_input -l hg38 --mp 1
Input file(s): /vf/users/denisovga/OpenCRAVAT/example_input
Genome assembly: hg38
Running converter...
Converter (converter) finished in 1.231s
Running gene mapper... finished in 5.209s
Running annotators...
annotator(s) finished in 1.027s
Running aggregator...
Variants finished in 0.147s
Genes finished in 0.115s
Samples finished in 0.145s
Tags finished in 0.253s
Indexing
variant base__coding finished in 0.015s
variant base__so finished in 0.014s
variant base__chrom finished in 0.009s
Running postaggregators...
Tag Sampler (tagsampler) finished in 0.076s
Finished normally. Runtime: 8.359s
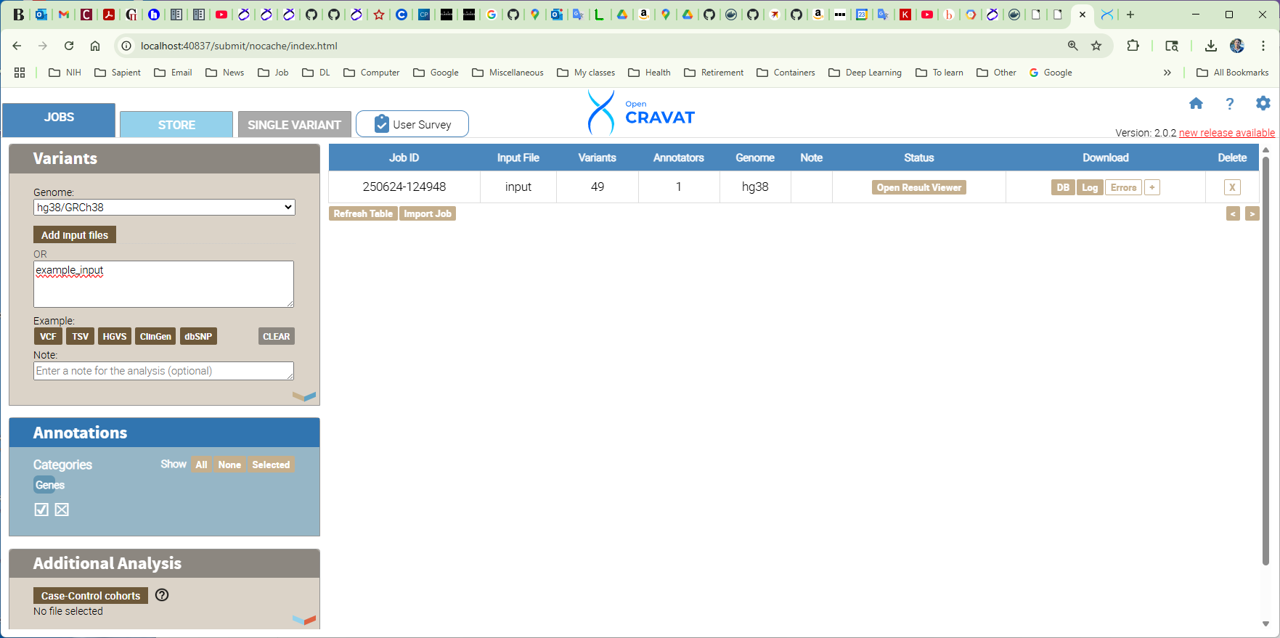
Once the job is finished, the following files wil be created: example_input.logIn particular, file example_input.sqlite is the sqlite database with the results.
example_input.sqlite
example_input.err
[user@cn2389 ~]$ oc gui --port $PORT1 example_input.sqlite
____ __________ ___ _ _____ ______
/ __ \____ ___ ____ / ____/ __ \/ | | / / |/_ __/
/ / / / __ \/ _ \/ __ \/ / / /_/ / /| | | / / /| | / /
/ /_/ / /_/ / __/ / / / /___/ _, _/ ___ | |/ / ___ |/ /
\____/ .___/\___/_/ /_/\____/_/ |_/_/ |_|___/_/ |_/_/
/_/
...
where $PORT1 is the tunneling port number you've got after allocating the interactive session. Store this port number and an id of the compute node you have been using, in this example node_id=cn2389ssh -t -L $PORT1:localhost:$PORT1 biowulf.nih.gov "ssh -L $PORT1:localhost:$PORT1 $node_id"where $PORT1 and $node_id should be replaced by the actual values you stored.
[user@cn2389 ~]$ exit salloc.exe: Relinquishing job allocation 46116226 [user@biowulf ~]$