Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S and Steinegger M.
ColabFold: Making protein folding accessible to all.
Nature Methods (2022) doi: 10.1038/s41592-022-01488-1.
PubMed |
PMC |
Journal
Module Name: colabfold (see the modules page for more information)
The MSA generation is a multithreaded process. Model generation uses GPU
Example files in $COLABFOLD_TEST_DATA
Reference data in /fdb/colabfold/
Currently including templates is not yet supported
colabfold_batch job scans *.done.txt under the output model directory and continues from where it is stopped/discrupted.
Resubmiting colabfold_batch with the same input and output directories will resume it, so users won't need to worry about hitting walltime.
Interactive job
Interactive jobs should be used for debugging, graphics, or applications that cannot be run as batch jobs.
Allocate an interactive session for
the generation of the multiple sequence applications (MSAs). This session
requires at least 128GB of memory. Note that
colabfold_search is optimized for running many query sequences in a
single job. Never run single sequences in the search part of
the analysis. It is inefficient and if you run many will strain the file
system. For example: Below we will create alignments for 17 polymerase
related proteins of the mimivirus genome which takes about 100 minutes.
Creating MSAs for all 979 proteins of the mimivirus genome take takes about 285
minutes or only 2.8x longer for 50x more proteins.
Note that colabfold_search
treats each protein in a fasta file as a separate monomer.
[user@biowulf]$ sinteractive --mem=128G --cpus-per-task=16 --gres=lscratch:100
salloc.exe: Pending job allocation 46116226
salloc.exe: job 46116226 queued and waiting for resources
salloc.exe: job 46116226 has been allocated resources
salloc.exe: Granted job allocation 46116226
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn3144 are ready for job
[user@cn3144]$ module load colabfold
[user@cn3144]$ cp $COLABFOLD_TEST_DATA/mimi_poly.fa .
[user@cn3144]$ colabfold_search --threads $SLURM_CPUS_PER_TASK \
mimi_poly.fa $COLABFOLD_DB mimi_poly_msa
[...much output...]
[user@cn3144]$ ls -lh mimi_poly_msa
total 66M
-rw-r--r-- 1 user group 17M Sep 22 16:14 0.a3m
-rw-r--r-- 1 user group 2.7M Sep 22 16:14 10.a3m
-rw-r--r-- 1 user group 92K Sep 22 16:14 11.a3m
-rw-r--r-- 1 user group 296K Sep 22 16:14 12.a3m
-rw-r--r-- 1 user group 33K Sep 22 16:14 13.a3m
-rw-r--r-- 1 user group 6.4M Sep 22 16:14 14.a3m
-rw-r--r-- 1 user group 42K Sep 22 16:14 15.a3m
-rw-r--r-- 1 user group 7.1K Sep 22 16:14 16.a3m
-rw-r--r-- 1 user group 23M Sep 22 16:14 1.a3m
-rw-r--r-- 1 user group 12M Sep 22 16:14 2.a3m
-rw-r--r-- 1 user group 797K Sep 22 16:14 3.a3m
-rw-r--r-- 1 user group 200K Sep 22 16:14 4.a3m
-rw-r--r-- 1 user group 580K Sep 22 16:14 5.a3m
-rw-r--r-- 1 user group 273K Sep 22 16:14 6.a3m
-rw-r--r-- 1 user group 160K Sep 22 16:14 7.a3m
-rw-r--r-- 1 user group 464K Sep 22 16:14 8.a3m
-rw-r--r-- 1 user group 2.6M Sep 22 16:14 9.a3m
[user@cn3144]$ exit
Now we can build the structure predictions for the 17 proteins above. This step requires
GPU. Each of the 17 proteins in this example is modeled as a monomer. See below for
multimer predictions. With the default settings on a A100 model generation for the 17
proteins takes approximately 4h
[user@biowulf]$ sinteractive --mem=48G --cpus-per-task=8 --gres=lscratch:100,gpu:a100:1
salloc.exe: Pending job allocation 46116227
salloc.exe: job 46116227 queued and waiting for resources
salloc.exe: job 46116227 has been allocated resources
salloc.exe: Granted job allocation 46116227
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn3144 are ready for job
[user@cn2144]$ module load colabfold
[user@cn2144]$ colabfold_batch --amber --use-gpu-relax \
mimi_poly_msa mimi_poly_models
2024-01-23 16:08:17,428 Running colabfold 1.5.5
2024-01-23 16:08:17,990 Unable to initialize backend 'rocm': NOT_FOUND: Could not find registered platform with name: "rocm". Available platform names are: CUDA Interpreter
2024-01-23 16:08:17,991 Unable to initialize backend 'tpu': module 'jaxlib.xla_extension' has no attribute 'get_tpu_client'
2024-01-23 16:08:22,823 Running on GPU
2024-01-23 16:08:24,124 Found 6 citations for tools or databases
2024-01-23 16:08:24,125 Query 1/3: 0 (length 1193)
2024-01-23 16:08:58,040 Padding length to 1203
warning: Linking two modules of different target triples: 'LLVMDialectModule' is 'nvptx64-nvidia-gpulibs' whereas '' is 'nvptx64-nvidia-cuda'
2024-01-23 16:11:24,669 alphafold2_ptm_model_1_seed_000 recycle=0 pLDDT=83.2 pTM=0.798
2024-01-23 16:13:27,900 alphafold2_ptm_model_1_seed_000 recycle=1 pLDDT=85.1 pTM=0.824 tol=1.09
[...snip...]
[user@cn2144]$ ls -lh mimi_poly_models | head
total 570M
-rw-r--r-- 1 user group 17M Sep 26 15:36 0.a3m
-rw-r--r-- 1 user group 142K Sep 26 16:19 0_coverage.png
-rw-r--r-- 1 user group 0 Sep 26 16:19 0.done.txt
-rw-r--r-- 1 user group 743K Sep 26 16:19 0_PAE.png
-rw-r--r-- 1 user group 196K Sep 26 16:19 0_plddt.png
-rw-r--r-- 1 user group 18M Sep 26 16:19 0_predicted_aligned_error_v1.json
-rw-r--r-- 1 user group 1.5M Sep 26 16:19 0_relaxed_rank_1_model_3.pdb
-rw-r--r-- 1 user group 1.5M Sep 26 16:19 0_relaxed_rank_2_model_4.pdb
-rw-r--r-- 1 user group 1.5M Sep 26 16:19 0_relaxed_rank_3_model_2.pdb
For each protein the output includes some diagnostic plots, the predicted alignment error
of the ranked models in json, as well as the ranked relaxed and unrelaxed models in PDB format.
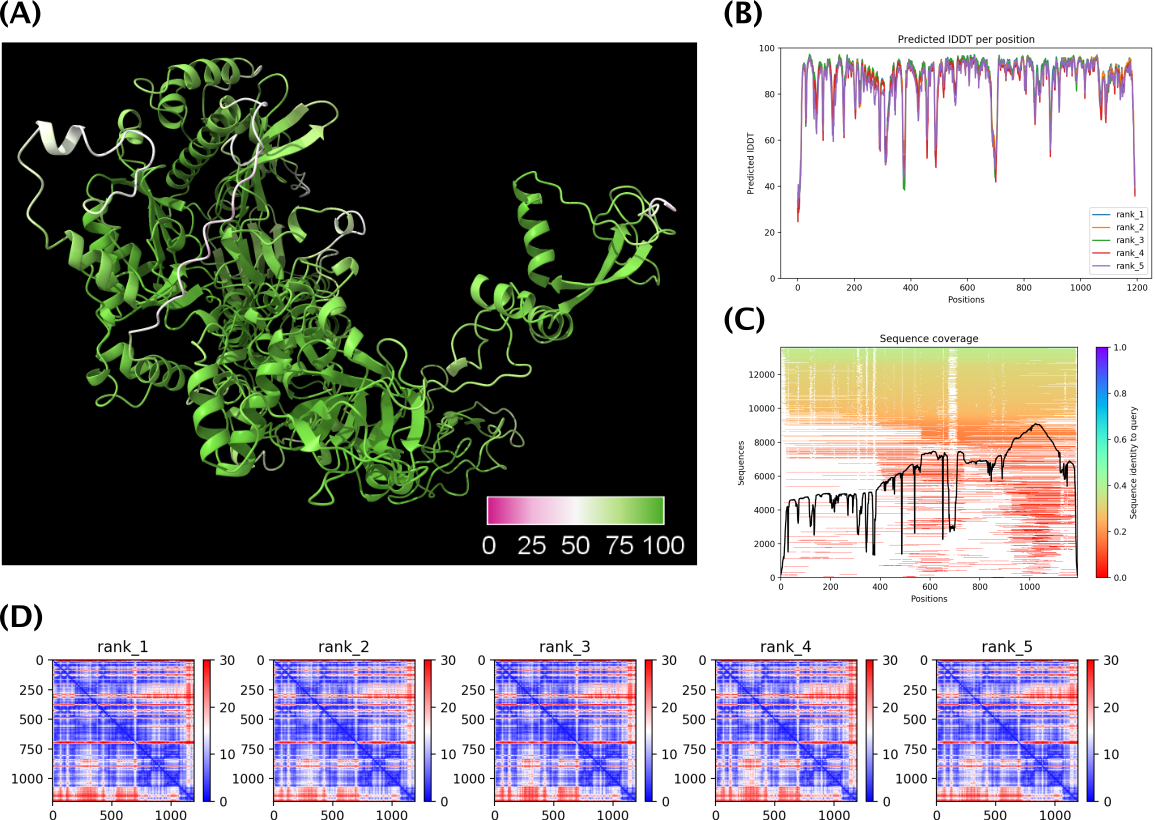
As an example below see the predicted best ranked model (rank 1) for YP_003986740 (DNA dependent
RNA polymerase subunit B) along with some of the diagnostic plots.
Figure 1. (A) The highest confidence
structure prediction (0_relaxed_rank_1_model_3.pdb) of protein
YP_003986740 colored by pLDDT. (B) pLDDT for all 5 models.
(C) Coverage of YP_003986740 in the MSA generated by colabfold_search
with mmseqs2. (D) Predicted aligned error (PAE) for all 5 models.
Commandline for generating two predictions for each of the 5 models per protein but only
relax the top 5 results
Now let's do an example with some custom templates. This example is somewhat contrived since
the templates are structures of the actual protein but it illustrates the process.