dyno is a meta package that installs several other packages
from the dynverse.
It comprises a set of R packages
to construct and interpret single-cell trajectories.
Allocate an interactive session and run the program. Sample session:
[user@biowulf ~]$ sinteractive --mem=12g -c4 --gres=lscratch:10 --tunnel
...
Created 1 generic SSH tunnel(s) from this compute node to
biowulf for your use at port numbers defined
in the $PORTn ($PORT1, ...) environment variables.
Please create a SSH tunnel from your workstation to these ports on biowulf.
On Linux/MacOS, open a terminal and run:
ssh -L 33521:localhost:33521 user@biowulf.nih.gov
For Windows instructions, see https://hpc.nih.gov/docs/tunneling
[user@cn0868 ~]$
Store the port number you get (in this case: 33521) and the id of the compute node you logged in to, in this case cn0868ssh -t -L 33521:localhost:33521 biowulf "ssh -L 33521:localhost:33521 cn0868"(make sure to use the actual port number you stored instead of 33521 and the actual node id number instead of cn0868).
[user@cn0868 ~]$ module load dyno
[+] Loading dyno 20220906 on cn0868
[+] Loading singularity 3.8.0 on cn0868
[user@cn0868 ~]$ R
> library(dyno)
Loading required package: dynfeature
Loading required package: dynguidelines
Loading required package: dynmethods
Loading required package: dynplot
Loading required package: dynwrap
> data("fibroblast_reprogramming_treutlein")
> dataset <- wrap_expression(counts = fibroblast_reprogramming_treutlein$counts, expression = fibroblast_reprogramming_treutlein$expression)
> guidelines <- guidelines_shiny(dataset, port=33521, launch.browser=F)
Loading required package: shiny
Listening on http://0.0.0.0:33521
Warning: All elements of `...` must be named.
Did you want `renderers = c(column_id, renderer, label, title, style, default, name, trajectory_type,
category_old, scaling_type)`?
Loading required namespace: akima
On your local system (PC or Mac), open a web browser or new tab on it, and navigate to: http://localhost:33521(make sure to use the port number you stored instead of 33521)
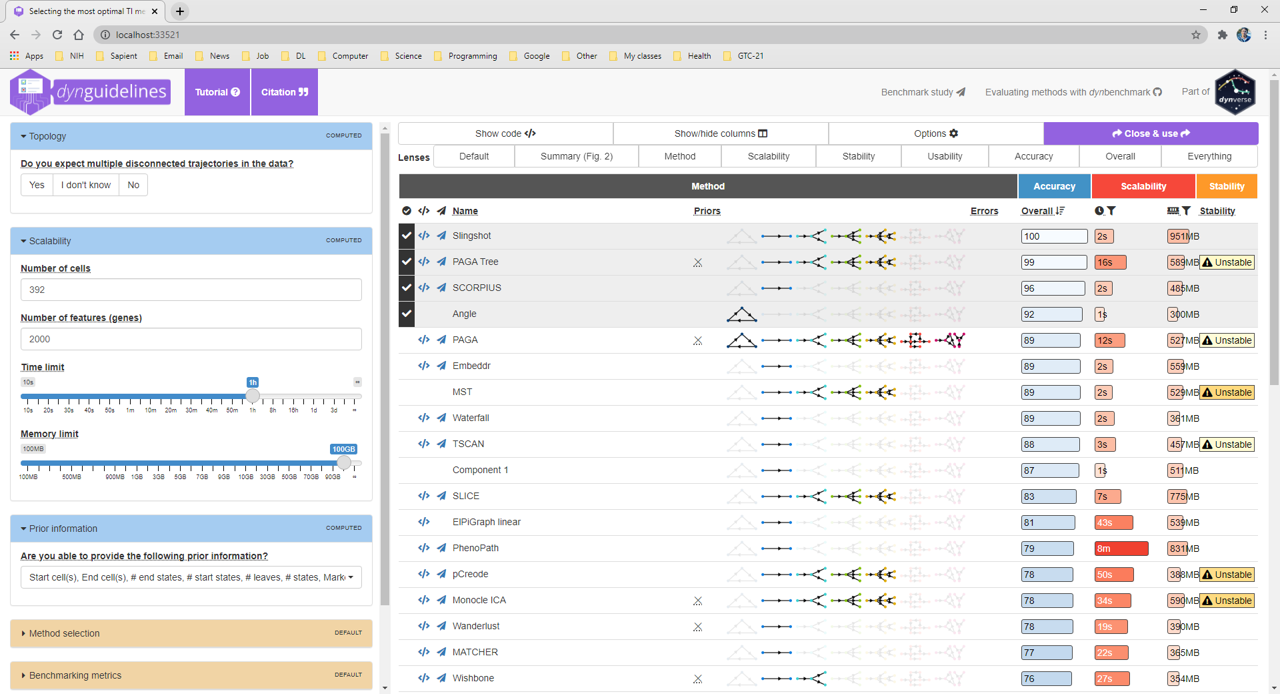
> methods_selected <- guidelines$methods_selected > methods_selected [1] "slingshot" "paga_tree" "scorpius" "angle" > model <- infer_trajectory(dataset, methods_selected[1]) Running singularity exec 'docker://dynverse/ti_slingshot:v1.0.3' echo hi ... Running /usr/local/current/singularity/3.8.0/bin/singularity exec \ --containall -B \ '/tmp/RtmppFshCi/file941a3d4b0be4/:/copy_mount,/tmp/RtmppFshCi/file941a8f3b018/tmp:/tmp2' \ 'docker://dynverse/ti_slingshot:v1.0.3' cp /code/definition.yml \ /copy_mount/ ...(the computation may take a few minutes)
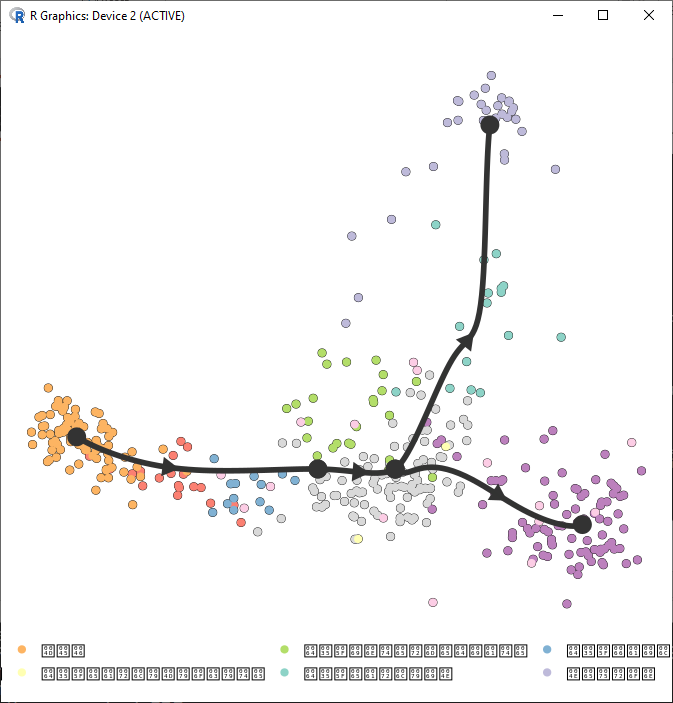
> library(magrittr) > model <- model %>% add_dimred(dyndimred::dimred_mds, expression_source = dataset$expression) > plot_dimred(model, expression_source = dataset$expression, grouping = fibroblast_reprogramming_treutlein$grouping ) Coloring by grouping Loading required namespace: RColorBrewer
> q() [user@cn0868 ~]$ exit exit salloc.exe: Relinquishing job allocation 49998864 [user@biowulf ~]$