
From the InterProScan home page:
InterProScan is the software package that allows sequences (protein and nucleic) to be scanned against InterPro's signatures. Signatures are predictive models, provided by several different databases, that make up the InterPro consortium.
$INTERPROSCAN_TEST_DATAInterProScan on our systems is used through a wrapper script that takes a subset of the native InterProScan options. The wrapper script then splits the input into chunks (if necessary) and sets up a slurm job array script to run interproscan in parallel on each chunk. The chunks are designed to run on the quick partition and make extensive use of the node-local lscratch disks.
Set up the environment and copy the example data (4303 proteins from the e. coli genome)
biowulf$ module load interproscan/5.72-103.0
[+] Loading java 12.0.1 ...
[+] Loading interproscan 5.72-103.0
biowulf$ cp ${INTERPROSCAN_TEST_DATA:-none}/ecoli_benchmark.fa .
biowulf$ grep -c '^>' ecoli_benchmark.fa
4303
biowulf$ interproscan --help
NAME
interproscan - set up and run an interproscan job on the cluster
SYNOPSIS
interproscan [interproscan options] <infile> <runname> <chunksize>
DESCRIPTION
This is a biowulf specific wrapper around interproscan.
It takes an input file and splits it up into chunks
that can run on the quick partition. It takes a subset of
interproscan options along with some arguments.
The <runname> is used to create a folder structure holding
potentially split input files and results for each chunk.
<chunksize> determines how many sequences are processed
by each subjob. This must be between 200 and 1000.
The following interpro options are the only options
supported by this wrapper:
-t,--seqtype <SEQUENCE-TYPE>
(n)ucleotide or (p)rotein. Default: p
-appl,--applications
Comma separated list of applications. Default: ALL
-dra,--disable-residue-annot
Optional, excludes sites from the XML, JSON output
-etra,--enable-tsv-residue-annot
Optional, includes sites in TSV output
-f,--formats <FORMAT-LIST>
Case-insensitive, comma separated list of output formats.
Supported formats are tsv, xml, json, gff3, svg (deprecated).
Defaults to tsv,json
-iprlookup,--iprlookup
Include InterPro annotation in output.
-goterms,--goterms
Include GO terms in output. Implies -iprlookup.
-pa,--pathways
Include pathway annotation in output. Implies -iprlookup
-ms,--minsize <MINIMUM-SIZE>
Minimum nt size of ORF to report. Will only be considered
if n is specified as a sequence type. Please be aware that
small values will lead to long runtimes.
Use --interproscan-help to see a full description of all the options
interproscan supports.
Setup and start the run
biowulf$ interproscan --goterms --pathways -f tsv,json ecoli_benchmark.fa test 600
Called from: /data/user/temp/interproscan
Interproscan root: /usr/local/apps/interproscan/5.72-103.0
Interproscan opts: --goterms --pathways -f tsv,json
Infile: ecoli_benchmark.fa
Chunksize: 600
Run name: test
Output dir /vf/users/wresch/temp/interproscan/test
sequences: 4303
jobs to run: 8
To submit your interproscan jobs do:
$ cd /data/user/temp/interproscan/test
$ sbatch interproscan.batch
The job script is set up to run on the quick partition with
a 4h walltime. Depending on your chunnk size that may not
be sufficient. If your jobs need more time please change
the batch script to use the norm partition and modify the
time limit.
biowulf$ ls -lh test
total 1.9M
-rw-rw-r-- 1 user group 253K Mar 14 10:05 ecoli_benchmark.fa.chunk0001
-rw-rw-r-- 1 user group 213K Mar 14 10:05 ecoli_benchmark.fa.chunk0002
-rw-rw-r-- 1 user group 247K Mar 14 10:05 ecoli_benchmark.fa.chunk0003
-rw-rw-r-- 1 user group 296K Mar 14 10:06 ecoli_benchmark.fa.chunk0004
-rw-rw-r-- 1 user group 275K Mar 14 10:06 ecoli_benchmark.fa.chunk0005
-rw-rw-r-- 1 user group 223K Mar 14 10:06 ecoli_benchmark.fa.chunk0006
-rw-rw-r-- 1 user group 260K Mar 14 10:06 ecoli_benchmark.fa.chunk0007
-rw-rw-r-- 1 user group 36K Mar 14 10:06 ecoli_benchmark.fa.chunk0008
-rw-rw-r-- 1 user group 950 Mar 14 10:06 interproscan.batch
drwxrwxr-x 2 user group 4.0K Mar 14 10:06 slurm_logs
biowulf$ cd test
biowulf$ cat interproscan.batch
#! /bin/bash
#SBATCH --mem=24g
#SBATCH --cpus-per-task=6
#SBATCH --gres=lscratch:50
#SBATCH --output=slurm_logs/slurm-%A_%a.out
#SBATCH --array=1-8
#SBATCH --time=4:00:00
#SBATCH --partition=quick
module load perl java/12.0.1 gnuplot || exit 1
fn=$(printf "%s.chunk%04i" "ecoli_benchmark.fa" $SLURM_ARRAY_TASK_ID)
cd /lscratch/$SLURM_JOB_ID
appdir=$PWD/$(basename /usr/local/apps/interproscan/5.52-86.0/interproscan_app)
cp "/data/user/test_data/interproscan/test/x/${fn}" .
cp -r /usr/local/apps/interproscan/5.52-86.0/interproscan_app .
# make panther use a local tempdir
sed -i 's:panther.temporary.file.directory=.*:panther.temporary.file.directory=/lscratch/$SLURM_JOB_ID/ttemp:' $appdir/interproscan.properties
# fix signalp script
sed -i -e "s:\$ENV{SIGNALP} =.*;:\$ENV{SIGNALP} = '$appdir/bin/signalp/4.1';:" -e "s:/var/tmp:/lscratch/$SLURM_JOB_ID/ttemp:" $appdir/bin/signalp/4.1/signalp
mkdir temp ttemp
# this will add the node local python first in the path and make it use
# local packages
export PATH=$appdir:$PATH
echo interproscan.sh --goterms --pathways -f tsv,json -T ./temp --disable-precalc -i "${fn}"
interproscan.sh --goterms --pathways -f tsv,json --highmem -T ./temp --disable-precalc -i "${fn}"
cp ${fn}.* /data/user/test_data/interproscan/test/x
biowulf$ sbatch interproscan.batch
biowulf$ jobload
JOBID TIME NODES CPUS THREADS LOAD MEMORY
Elapsed / Wall Alloc Active Used / Alloc
35660753_8 00:01:12 / 04:00:00 cn2232 8 9 112% 2.0 / 10.0 GB
35660753_1 00:01:12 / 04:00:00 cn2311 8 9 112% 1.7 / 10.0 GB
35660753_2 00:01:12 / 04:00:00 cn2314 8 8 100% 1.6 / 10.0 GB
35660753_3 00:01:12 / 04:00:00 cn2276 8 7 88% 1.6 / 10.0 GB
35660753_4 00:01:12 / 04:00:00 cn2277 8 11 138% 1.6 / 10.0 GB
35660753_5 00:01:12 / 04:00:00 cn2477 8 9 112% 1.5 / 10.0 GB
35660753_6 00:01:12 / 04:00:00 cn2478 8 8 100% 1.5 / 10.0 GB
35660753_7 00:01:12 / 04:00:00 cn2231 8 9 112% 1.7 / 10.0 GB
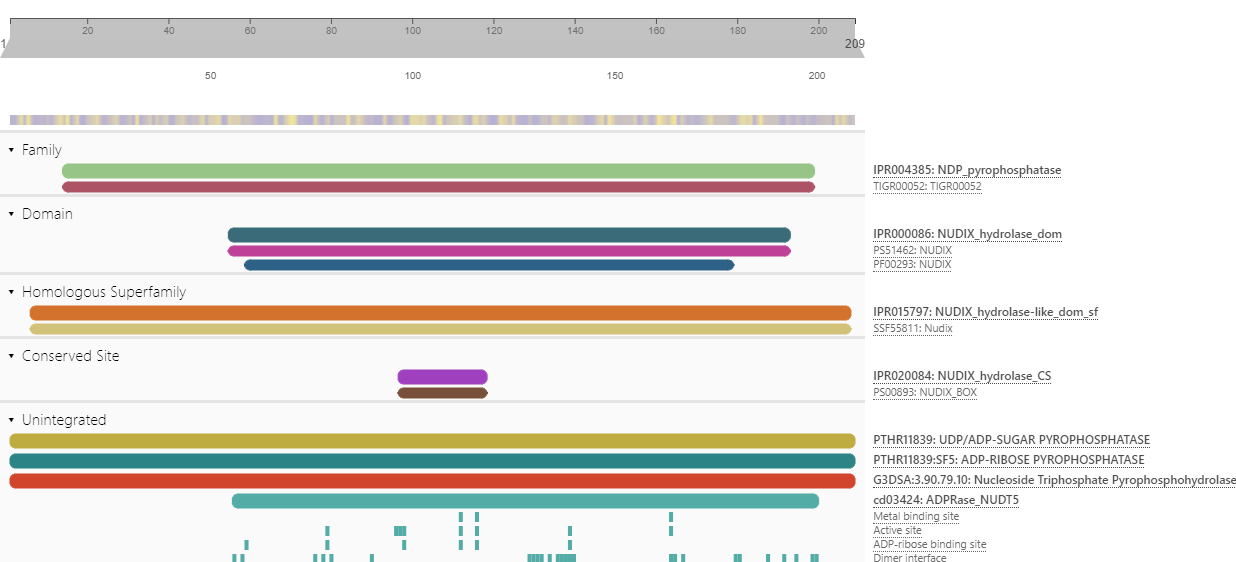
In this particular example the run takes about 40m. The following image is an example of what the SVG output looked like for interproscan versions before 5.52-86.0
And here is example output for a different protein obtained by uploading json output from 5.52-86.0 to the interproscan page
Also available is a convert script that calls interproscan -mode convert and is
used to convert XML output files to other formats