Description
LAMMPS is a classical molecular dynamics code, and an acronym for Large-scale Atomic/Molecular Massively Parallel Simulator. It runs on a variety of different computer systems, including single processor systems, distributed-memory machines with MPI, and GPU and Xeon Phi systems. LAMMPS is open source software, released under the GNU General Public License.
LAMMPS can be accesed via modules. To see the modules available, type
module avail lammps
To select a module, type
module load lammps/[ver]
where [ver] is the version of choice. Currently, versions 30Jul16 and 31Mar17 are installed.
Installed YES: package ASPHERE Installed YES: package BODY Installed YES: package CLASS2 Installed YES: package COLLOID Installed YES: package COMPRESS Installed YES: package CORESHELL Installed YES: package DIPOLE Installed NO: package GPU Installed YES: package GRANULAR Installed NO: package KIM Installed NO: package KOKKOS Installed YES: package KSPACE Installed YES: package MANYBODY Installed YES: package MC Installed NO: package MEAM Installed YES: package MISC Installed YES: package MOLECULE Installed NO: package MPIIO Installed YES: package OPT Installed YES: package PERI Installed NO: package POEMS Installed YES: package PYTHON Installed YES: package QEQ Installed NO: package REAX Installed YES: package REPLICA Installed YES: package RIGID Installed YES: package SHOCK Installed YES: package SNAP Installed YES: package SRD Installed NO: package VORONOI
All executables have the following packages:
Installed YES: package ASPHERE Installed YES: package BODY Installed YES: package CLASS2 Installed YES: package COLLOID Installed YES: package COMPRESS Installed YES: package CORESHELL Installed YES: package DIPOLE Installed NO: package GPU Installed YES: package GRANULAR Installed NO: package KIM Installed NO: package KOKKOS Installed YES: package KSPACE Installed YES: package MANYBODY Installed YES: package MC Installed NO: package MEAM Installed YES: package MISC Installed YES: package MOLECULE Installed NO: package MPIIO Installed NO: package MSCG Installed YES: package OPT Installed YES: package PERI Installed NO: package POEMS Installed YES: package PYTHON Installed YES: package QEQ Installed NO: package REAX Installed YES: package REPLICA Installed YES: package RIGID Installed YES: package SHOCK Installed YES: package SNAP Installed YES: package SRD Installed NO: package VORONOI
In addition, the lmp_icc_serial.user and lmp_icc_openmpi.user binaries have the following USER packages installed:
Installed NO: package USER-ATC Installed NO: package USER-AWPMD Installed YES: package USER-CG-CMM Installed YES: package USER-CGDNA Installed NO: package USER-COLVARS Installed YES: package USER-DIFFRACTION Installed YES: package USER-DPD Installed YES: package USER-DRUDE Installed YES: package USER-EFF Installed YES: package USER-FEP Installed NO: package USER-H5MD Installed YES: package USER-INTEL Installed NO: package USER-LB Installed YES: package USER-MANIFOLD Installed YES: package USER-MGPT Installed YES: package USER-MISC Installed YES: package USER-MOLFILE Installed YES: package USER-NC-DUMP Installed YES: package USER-OMP Installed NO: package USER-PHONON Installed NO: package USER-QMMM Installed YES: package USER-QTB Installed NO: package USER-QUIP Installed YES: package USER-REAXC Installed NO: package USER-SMD Installed YES: package USER-SMTBQ Installed YES: package USER-SPH Installed YES: package USER-TALLY Installed NO: package USER-VTK
Installed YES: package ASPHERE Installed YES: package BODY Installed YES: package CLASS2 Installed YES: package COLLOID Installed YES: package COMPRESS Installed YES: package CORESHELL Installed YES: package DIPOLE Installed NO: package GPU Installed YES: package GRANULAR Installed NO: package KIM Installed NO: package KOKKOS Installed YES: package KSPACE Installed NO: package LATTE Installed YES: package MANYBODY Installed YES: package MC Installed NO: package MEAM Installed YES: package MISC Installed YES: package MOLECULE Installed NO: package MPIIO Installed NO: package MSCG Installed YES: package OPT Installed YES: package PERI Installed NO: package POEMS Installed YES: package PYTHON Installed YES: package QEQ Installed NO: package REAX Installed YES: package REPLICA Installed YES: package RIGID Installed YES: package SHOCK Installed YES: package SNAP Installed YES: package SRD Installed NO: package VORONOI Installed NO: package USER-ATC Installed NO: package USER-AWPMD Installed YES: package USER-CGDNA Installed YES: package USER-CGSDK Installed YES: package USER-COLVARS Installed YES: package USER-DIFFRACTION Installed YES: package USER-DPD Installed YES: package USER-DRUDE Installed YES: package USER-EFF Installed YES: package USER-FEP Installed NO: package USER-H5MD Installed NO: package USER-INTEL Installed NO: package USER-LB Installed YES: package USER-MANIFOLD Installed YES: package USER-MEAMC Installed YES: package USER-MESO Installed YES: package USER-MGPT Installed YES: package USER-MISC Installed YES: package USER-MOLFILE Installed YES: package USER-NETCDF Installed YES: package USER-OMP Installed NO: package USER-PHONON Installed NO: package USER-QMMM Installed YES: package USER-QTB Installed NO: package USER-QUIP Installed YES: package USER-REAXC Installed NO: package USER-SMD Installed YES: package USER-SMTBQ Installed YES: package USER-SPH Installed YES: package USER-TALLY Installed YES: package USER-UEF Installed NO: package USER-VTK
IMPORTANT NOTE: The 29Oct20 version was built with gcc/g++ instead of the Intel coompilers. Therefore, the executable names are lmp_g++_serial and lmp_g++_openmpi. The Following packages are installed in both versions:
Installed YES: package ASPHERE Installed YES: package BODY Installed YES: package CLASS2 Installed YES: package COLLOID Installed YES: package COMPRESS Installed YES: package CORESHELL Installed YES: package DIPOLE Installed NO: package GPU Installed YES: package GRANULAR Installed NO: package KIM Installed NO: package KOKKOS Installed YES: package KSPACE Installed NO: package LATTE Installed YES: package MANYBODY Installed YES: package MC Installed NO: package MESSAGE Installed YES: package MISC Installed NO: package MLIAP Installed YES: package MOLECULE Installed NO: package MPIIO Installed NO: package MSCG Installed YES: package OPT Installed YES: package PERI Installed NO: package POEMS Installed YES: package PYTHON Installed YES: package QEQ Installed YES: package REPLICA Installed YES: package RIGID Installed YES: package SHOCK Installed YES: package SNAP Installed NO: package SPIN Installed NO: package SRD Installed NO: package VORONOI Installed NO: package USER-ADIOS Installed NO: package USER-ATC Installed NO: package USER-AWPMD Installed NO: package USER-BOCS Installed YES: package USER-CGDNA Installed YES: package USER-CGSDK Installed YES: package USER-COLVARS Installed YES: package USER-DIFFRACTION Installed YES: package USER-DPD Installed YES: package USER-DRUDE Installed YES: package USER-EFF Installed YES: package USER-FEP Installed NO: package USER-H5MD Installed NO: package USER-INTEL Installed NO: package USER-LB Installed YES: package USER-MANIFOLD Installed YES: package USER-MEAMC Installed NO: package USER-MESODPD Installed NO: package USER-MESONT Installed YES: package USER-MGPT Installed YES: package USER-MISC Installed NO: package USER-MOFFF Installed YES: package USER-MOLFILE Installed YES: package USER-NETCDF Installed YES: package USER-OMP Installed NO: package USER-PHONON Installed NO: package USER-PLUMED Installed NO: package USER-PTM Installed NO: package USER-QMMM Installed YES: package USER-QTB Installed NO: package USER-QUIP Installed NO: package USER-REACTION Installed YES: package USER-REAXC Installed NO: package USER-SCAFACOS Installed NO: package USER-SMD Installed YES: package USER-SMTBQ Installed NO: package USER-SDPD Installed YES: package USER-SPH Installed YES: package USER-TALLY Installed YES: package USER-UEF Installed NO: package USER-VTK Installed NO: package USER-YAFF
LAMMPS is a parallel, computationally intensive program. It is therefore not permitted to be used on Helix.
Create a batch input file (e.g. run_lammps.sh), which uses the input file 'job.in'. For example:
#!/bin/bash #SBATCH --partition=multinode #SBATCH --ntasks-per-core=1 #SBATCH --time=12:00:00 #SBATCH --exclusive module load lammps/30Jul16 `which mpirun` lmp_icc_openmpi < job.in
Submit this job using the Slurm sbatch command.
sbatch --ntasks=64 run_lammps.sh
NOTE 1: You will need to adjust this script to meet your own needs. This script is set to run on the multinode partition using 64 cores (4 nodes, each of which have 16 cores or 128 CPUs - however, LAMMPS, being floating-point operation heavy, does not make use of the second CPU on a core, therefore, we use --ntasks-per-core=1). You should benchmark your own system to find an optimal number of nodes to run on.
NOTE 2: LAMMPS will also run on the "norm" partition, if you do not need to use more than a single node (16 cores = 32 CPUs). However, the version currently compiled will not run on the b1 nodes.
NOTE 3: We do not currently have a GPU-enabled version of LAMMPS available. Please contact staff@hpc.nih.gov if you are interested in using LAMMPS on GPUs, and we can install the GPU-enabled version.
NOTE 4: If using version 29Oct20, the name of the executable to pass to the mpirun command is lmp_g++_openmpi.
A sample LAMMPS input file is given below, for a system with a solvated GroEL chaperonin protein. This input file instructs LAMMPS to run 10,000 steps of molecular dynamics on the system using the CHARMM force field.
# Created by charmm2lammps v1.8.3 on Mon Oct 10 14:48:11 EDT 2016 units real neigh_modify delay 2 every 1 atom_style full bond_style harmonic angle_style charmm dihedral_style charmm improper_style harmonic pair_style lj/charmm/coul/long 10 12 pair_modify mix arithmetic kspace_style pppm 1e-4 read_data groel.data special_bonds charmm thermo 100 thermo_style multi timestep 1.0 fix 1 all nve fix 2 all shake 1e-6 500 0 m 1.0 a 127 velocity all create 0.0 12345678 dist uniform log log.groel run 10000
Note that the exact force field parameters are generated from the CHARMM force field parameter files using the charmm2lammps utility, which is described further below.
Specifying a homogenous set of nodes
The 'multinode' partition, to which all jobs that require more than a single node must be submitted, is heterogenous. For efficient parallel jobs, you need to ensure that you request nodes of a single CPU type. For example, at the time of writing this webpage, the 'freen' command displays:
biowulf% freen ... multinode 65/466 3640/26096 28 56 248g 400g cpu56,core28,g256,ssd400,x2695,ibfdr multinode 4/190 128/6080 16 32 60g 800g cpu32,core16,g64,ssd800,x2650,ibfdr multinode 312/539 17646/30184 28 56 250g 800g cpu56,core28,g256,ssd800,x2680,ibfdr ...These lines indicate that there are 3 kinds of nodes in the multinode partition. You should submit your job exclusively to one kind of node by specifying --constraint=x2695, --constraint=x2650, or --constrant=x2680 as in the examples below.
LAMMPS is not an interactive program, but in principle it can be run interactively for short test/debug runs. An example of an interactive session might look like:
biowulf$ sintetactive --constraint=x2650 salloc.exe: Pending job allocation NNNN salloc.exe: job NNNN queued and waiting for resources salloc.exe: job NNNN has been allocated resources salloc.exe: Granted job allocation 24912014 salloc.exe: Waiting for resource configuration salloc.exe: Nodes cnXXXX are ready for job srun: error: x11: no local DISPLAY defined, skipping cnXXXX$ module load lammps [+] Loading Intel 2015.1.133 Compilers ... [+] Loading openmpi 1.10.0 for Intel 2015.1.133 [+] Loading lammps 30Jul16 ... cnXXXX$ lmp_icc_serial < input.file ... lots of LAMMPS output ... cnXXXX$ exit biowulf$
NOTE: If using version 29Oct20, the correct executable name is lmp_g++_serial.
LAMMPS has a utility that can be used to convert CHARMM PSF and coordinate (or PDB) files to input suitable for LAMMPS itself. These tools are in /usr/local/apps/lammps/30Jul16/tools/ch2lmp (there is also an amber2lmp directory that contains tools for converting AMBER input). Below, I give an example of converting a system.
$ perl /usr/local/apps/lammps/30Jul16/tools/ch2lmp/charmm2lammps.pl all27_prot_na groel charmm2lammps v1.8.3 (c)2016 Info: using groel.pdb instead of groel.crd Info: lx not set: will use extremes Info: ly not set: will use extremes Info: lz not set: will use extremes Info: creating PSF index Info: converting atoms Info: converting bonds Info: converting angles Info: converting dihedrals Info: converting impropers Info: conversion complete
Please note:
Two benchmark systems were run - a Rhodopsin protein in a lipid bilayer (32,000 atoms total) using the NPT statistical ensemble and a GroEL chaperonin in explicit water (~ 468,000 atoms total). The Rhodopsin system was taken from the included LAMMPS benchmarks (/usr/local/apps/lammps/30Jul16/bench), while the GroEL was generated from existing CHARMM input files using charmm2lammps - described above. Note that the Rhodopsin benchmark was run with 2 femtosecond time steps and GroEL was run with 1 femtosecond time steps. The benchmarks were run on Biowulf InfiniBand nodes.
The timings given below are for 10,000 molecular dynamics steps of each system, running on 4-256 cores.
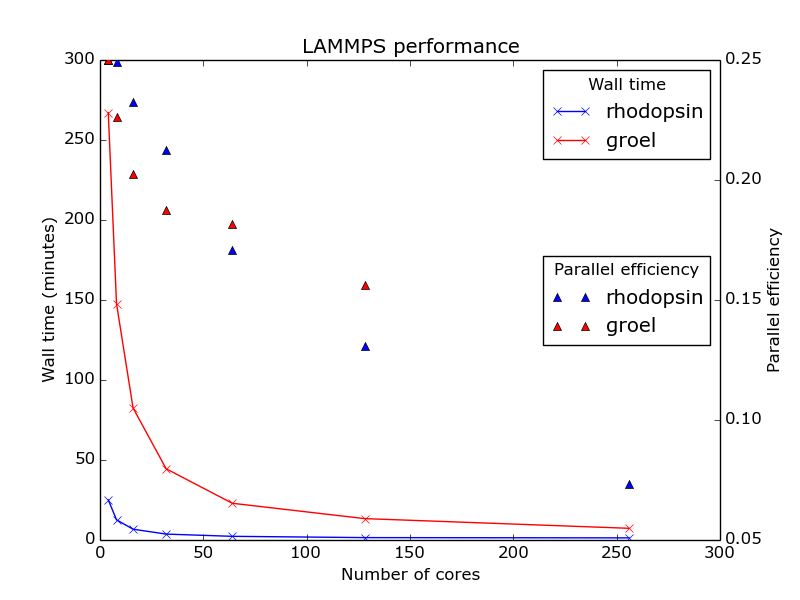
| Number of cores | 10,000 step time for Rhodopsin (seconds) | 10,000 step time for GroEL (seconds) |
|---|---|---|
| 4 | 1506.21 (1.147 ns/day) | 16028.16 (0.054 ns/day) |
| 8 | 756.18 (2.285 ns/day) | 8855.12 (0.098 ns/day) |
| 16 | 404.95 (4.267 ns/day) | 4951.71 (0.174 ns/day) |
| 32 | 221.62 (7.797 ns/day) | 2671.87 (0.323 ns/day) |
| 64 | 137.81 (12.539 ns/day) | 1377.71 (0.627 ns/day) |
| 128 | 89.88 (19.227 ns/day) | 802.22 (1.077 ns/day) |
| 256 | 80.08 (21.577 ns/day) | 438.87 (1.969 ns/day) |