From the LASER home page:
LASER is a program to estimate individual ancestry by directly analyzing shotgun sequence reads without calling genotypes. LASER uses principal components analysis (PCA) and Procrustes analysis to analyze sequence reads of each sample and place the sample into a reference PCA space constructed using genotypes of a set of reference individuals. With an appropriate reference panel, the estimated coordinates of the sequence samples reflect their ancestral background and can be used to correct for population stratification in association studies. LASER can accurately estimate ancestry even with modest amounts of data, such as the off-target sequence data generated by targeted sequencing experiments.
In version 2.0 or later, the software package includes a new program TRACE for tracing an individual's genetic ancestry based on genotype data. TRACE follows the same analysis framework as LASER and can accurately place study samples into a reference ancestry space using a relatively small number of genotypes. When using the same reference panel, LASER and TRACE can place sequenced and genotyped samples into the same ancestry space.
LASER can also perform standard PCA on genotype data to explore population structure and to create the reference ancestry space. Different options to compute PC scores and PC loadings have been implemented in the LASER program (version 2.01 or later).
There may be multiple versions of laser available. An easy way of selecting the version is to use modules. To see the modules available, type
module avail laser
To select a module use
module load laser/[version]
where [version] is the version of choice.
$PATH$LASET_TEST_DATAAllocate an interactive session with sinteractive and set up your environment
biowulf$ sinteractive --mem=10g node$ module load laser [+] Loading laser 2.04 node$ exit biowulf$
First, we will use ~9k SNP loci on chr22 of 700 individuals from
the Human Genome Diversity Panel (HGDP) as a reference set to calculate
PCA coordinates. laser uses a combination of a configuration
file and command line options:
node$ cp -r $LASER_TEST_DATA/HapMap_6_chr22.* $LASER_TEST_DATA/HGDP_700* . node$ ls -lh total 15M -rw-r--r-- 1 user group 348K Jul 10 16:31 HapMap_6_chr22.seq -rw-r--r-- 1 user group 243K Jul 10 16:31 HapMap_6_chr22.site -rw-r--r-- 1 user group 13M Jul 10 16:31 HGDP_700_chr22.geno -rw-r--r-- 1 user group 243K Jul 10 16:31 HGDP_700_chr22.site node$ cat > test.config <<EOF GENO_FILE HGDP_700_chr22.geno SEQ_FILE HapMap_6_chr22.seq DIM 8 EOF node$ laser -p ./test.conf -pca 1 -o HGDP_700_chr22 ==================================================================== ==== LASER: Locating Ancestry from SEquence Reads ==== ==== Version 2.04, Last updated on Jan/11/2017 ==== ==== (C) 2013-2017 Chaolong Wang, GNU GPL v3.0 ==== ==================================================================== Started at: Mon Jul 10 16:41:20 2017 6 individuals in the SEQ_FILE. 9608 loci in the SEQ_FILE. 700 individuals in the GENO_FILE. 9608 loci in the GENO_FILE. Parameter values used in execution: ------------------------------------------------- GENO_FILE (-g) HGDP_700_chr22.geno DIM (-k) 8 OUT_PREFIX (-o) HGDP_700_chr22 CHECK_FORMAT (-fmt) 10 PCA_MODE (-pca) 1 RANDOM_SEED (-seed) 0 NUM_THREADS (-nt) 8 ------------------------------------------------- Mon Jul 10 16:41:20 2017 Checking data format ... GENO_FILE: OK. SEQ_FILE: OK. COORD_FILE: not specified. Mon Jul 10 16:41:20 2017 Identify 9608 loci shared by SEQ_FILE and GENO_FILE. The analysis will base on the remaining 9608 loci. Mon Jul 10 16:41:20 2017 Reading reference genotypes ... Mon Jul 10 16:41:21 2017 Performing PCA on reference genotypes ... Genetic relationship matrix is output to 'HGDP_700_chr22.RefPC.grm'. Z scores of reference individuals are output to 'HGDP_700_chr22.RefPC.zscore'. Reference PCA coordinates are output to 'HGDP_700_chr22.RefPC.coord'. Variances explained by PCs are output to 'HGDP_700_chr22.RefPC.var'. Finished at: Mon Jul 10 16:41:23 2017 Total wall clock time: 3.02811 seconds. ==================================================================== node$ head HGDP_700_chr22.RefPC.coord popID indivID PC1 PC2 PC3 PC4 PC5 PC6 PC7 PC8 Brahui HGDP00005 2.81178 14.9014 -1.47298 -3.51307 19.0424 -1.46033 10.8402 0.777612 Brahui HGDP00007 -0.0477848 16.4082 1.4118 -4.20778 15.3486 -5.65236 7.55021 10.5837 Brahui HGDP00009 -0.567587 14.5716 2.67543 -4.94805 6.51882 1.95237 2.0824 0.0295942 [...snip...]
The reference coordinate file does not have to be calculated separately
as we've done here. However, if the analysis of unknown samples is to be done
more than once or if samples are to be split, it is computationally more
efficient. Next, we can analyze some unknown samples. We add the name
of the coord file to the config file and omit the -pca
to do so. In addition, since this is a stochastic process we will run it
three times and take the averate coordinates (-r 3):
node$ echo "COORD_FILE HGDP_700_chr22.RefPC.coord >> test.conf node$ laser -p test.conf -r 3 -o test ==================================================================== ==== LASER: Locating Ancestry from SEquence Reads ==== ==== Version 2.04, Last updated on Jan/11/2017 ==== ==== (C) 2013-2017 Chaolong Wang, GNU GPL v3.0 ==== ==================================================================== Started at: Mon Jul 10 16:55:24 2017 6 individuals in the SEQ_FILE. 9608 loci in the SEQ_FILE. 700 individuals in the GENO_FILE. 9608 loci in the GENO_FILE. 700 individuals in the COORD_FILE. 8 PCs in the COORD_FILE. [...snip...] Mon Jul 10 16:55:25 2017 Reading reference PCA coordinates ... Mon Jul 10 16:55:25 2017 Analyzing sequence samples ... Results for the sequence samples are output to: 'test.SeqPC.coord' (mean across 3 repeated runs) 'test.SeqPC.coord.sd' (standard deviation across 3 repeated runs) Finished at: Mon Jul 10 16:55:37 2017 Total wall clock time: 13.5122 seconds. ====================================================================
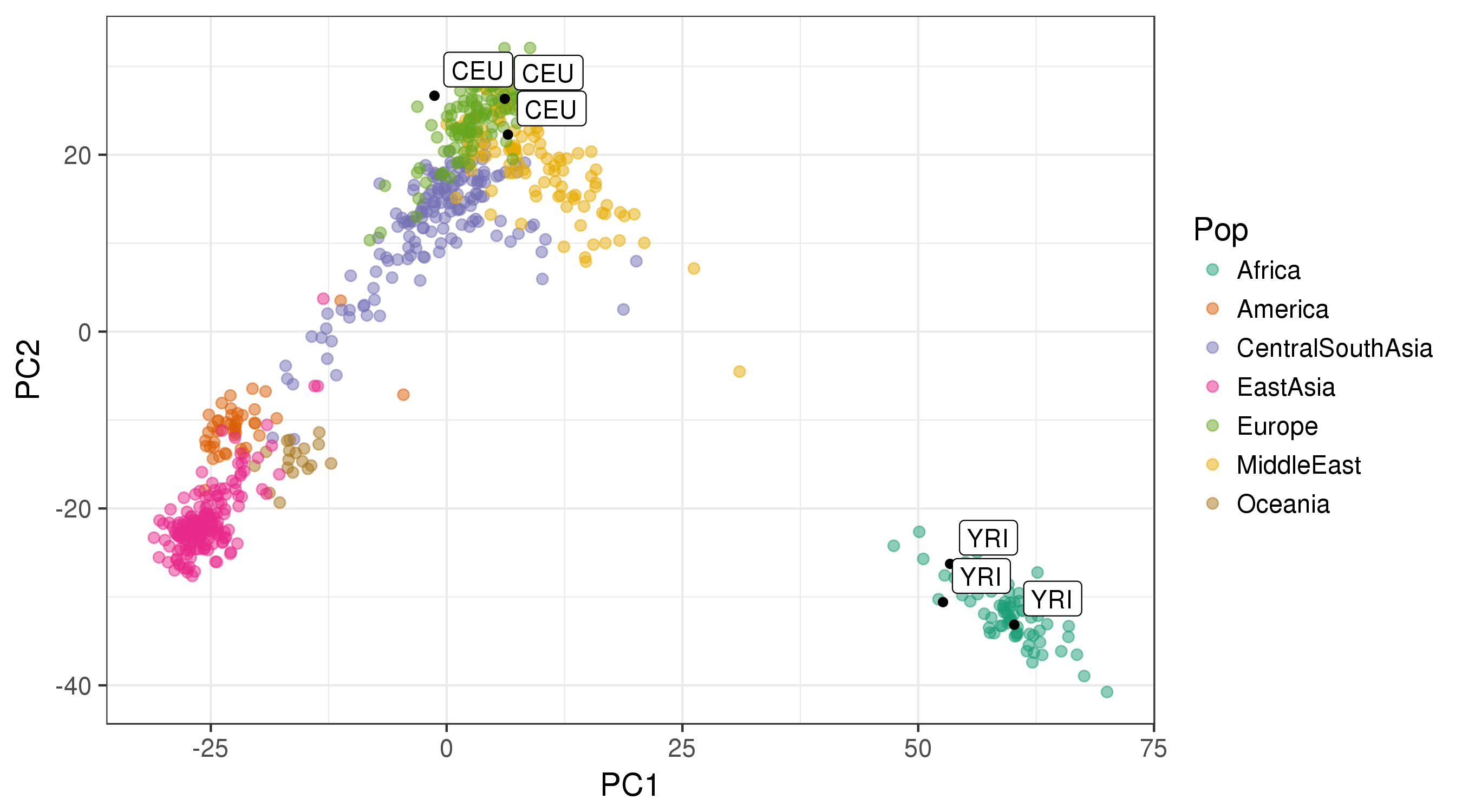
The graph below is an illustration of the results (plotting the 6 unknown samples on top of the colored reference data):
There are two utilities to create the genotype and site files:
| Tool | Descriptiont |
|---|---|
| vcf2geno | convert a VCF file to a genotype and site file |
| pileup2seq.py | used together with samtools pileup to produce .seq file |
Create a batch script similar to the following example:
#! /bin/bash # this file is laser.batch module load laser/2.04 || exit 1 # create the config file and run the PCA on the reference panel echo "GENO_FILE HGDP_700_chr22.geno" > test.conf echo "SEQ_FILE HapMap_6_chr22.seq" >> test.conf echo "DIM 8" >> test.conf laser -p test.conf -pca 1 -o HGDP_700_chr22 # now run the analysis in two parallel chunks (Sequences 1-3 in the first # chunk; sequences 4-6 in the second chunk) echo "COORD_FILE HGDP_700_chr22.RefPC.coord" >> test.conf laser -p test.conf -r3 -x 1 -y 3 -o out.1-3 & laser -p test.conf -r3 -x 4 -y 6 -o out.4-6 & wait
Submit to the queue with sbatch:
biowulf$ sbatch --mem=10g --cpus-per-task=2 laser.batch