Snakemake aims to reduce the complexity of creating workflows by providing a fast and comfortable execution environment, together with a clean and modern domain specific specification language (DSL) in python style.
You can use our snakemake profile which takes generic resource keys/values and automatically determines partition assignment for jobs. It also uses a low impact method of querying for jobs.
For a general introduction on how to use snakemake please read through the official Documentation and Tutorial or any of the other materials mentioned above.
In addition, you can have a look at a set of exercises on our GitHub site.
Allocate an interactive session and use as described below. If all tasks are submitted as cluster jobs this session may only require 2 CPUs. If some or all of the rules are run locally (i.e. within the interactive session) please adjust the resource requirements accordingly. In the example above there are at least 3 rules that are going to be run locally so we will request 8 CPUs.
[user@biowulf]$ sinteractive --mem=6g --cpus-per-task=8 salloc.exe: Pending job allocation 46116226 salloc.exe: job 46116226 queued and waiting for resources salloc.exe: job 46116226 has been allocated resources salloc.exe: Granted job allocation 46116226 salloc.exe: Waiting for resource configuration salloc.exe: Nodes cn3144 are ready for job [user@cn3144 ~]$ module load snakemake [user@cn3144 ~]$ # for the local rules [user@cn3144 ~]$ module load samtools/1.3.1 [user@cn3144 ~]$ cd /path/to/snakefile [user@cn3144 ~]$ ls -lh -rw-rw-r-- 1 user group 4.0K May 12 2015 config.yaml -rw-rw-r-- 1 user group 4.8K Jan 30 15:06 Snakefile
Run the pipeline locally (i.e. each task would be run as part
of this interactive session). Note that snakemake
will by default assume that your pipeline is in a file called 'Snakefile'. If
that is not the case, provide the filename with '-s SNAKEFILENAME'.
This would take a long time:
[user@cn3144 ~]$ snakemake -pr --keep-going -j $SLURM_CPUS_PER_TASK all
Provided cores: 8
Job counts:
count jobs
14 align
1 all
14 clean_fastq
14 fastqc
6 find_broad_peaks
8 find_narrow_peaks
14 flagstat_bam
14 index_bam
85
[...snip...]
To submit subjobs that are not marked as localrules to the cluster it
is necessary to provide an sbatch template string using variables from the
Snakefile or the cluster configuration file. Please use --max-jobs-per-second
and --max-status-checks-per-second in your command line or profile to limit
the number of calls to slurm:
[user@cn3144 ~]$ sbcmd="sbatch --cpus-per-task={threads} --mem={resources.mem_mb}"
[user@cn3144 ~]$ sbcmd+=" --time={resources.runtime} --partition={resources.partition}"
[user@cn3144 ~]$ sbcmd+=" --out=logs/slurm-%j.out"
[user@cn3144 ~]$ snakemake -pr --keep-going --local-cores $SLURM_CPUS_PER_TASK \
--jobs 10 --cluster "$sbcmd" \
--max-jobs-per-second 1 --max-status-checks-per-second 0.01 \
--latency-wait 120 all
Provided cluster nodes: 10
Job counts:
count jobs
14 align
1 all
14 clean_fastq
14 fastqc
6 find_broad_peaks
8 find_narrow_peaks
14 flagstat_bam
14 index_bam
85
[...snip...]
[user@cn3144 ~]$ exit
salloc.exe: Relinquishing job allocation 46116226
[user@biowulf ~]$
Note that --latency-wait 120 is required for pipelines that
submit cluster jobs as output files generated on other nodes may not
become visible to the parental snakemake job until after some delay.
In this usage mode the main snakemake job is itself submitted as a batch job. It is still possible to either run all the rules locally as part of the single job or have the main job submit each (non-local) rule as another batch job as described above. The example below uses the latter pattern.
Create a batch input file (e.g. snakemake.sh) similar to the following:
#! /bin/bash
# this file is snakemake.sh
module load snakemake samtools || exit 1
sbcmd="sbatch --cpus-per-task={threads} --mem={resources.mem_mb}"
sbcmd+=" --time={resources.runtime} --partition={resources.partition}"
sbcmd+=" --out=logs/slurm-%j.out"
snakemake -pr --keep-going --local-cores $SLURM_CPUS_PER_TASK \
--jobs 10 --cluster "$sbcmd" \
--latency-wait 120 all
Submit this job using the Slurm sbatch command.
sbatch --cpus-per-task=2 --mem=8g snakemake.sh
For pipelines with many short running jobs we highly recomment you explore job groups to combine multiple tasks into single batch jobs. Here is a trivial example pipeline where for each of 5 input "samples" three jobs are executed before results are summarized. Resources are specified in the Snakefile directly:
ids = [1, 2, 3, 4, 5]
localrules: all
rule all:
input: expand("stage3/{id}.3", id=ids)
output: "output/summary"
shell: "cat {input} > {output}"
rule stage1:
input: "input/{id}"
output: "stage1/{id}.1"
threads: 2
resources: mem_mb=1024, runtime=5
shell: "cp {input} {output}"
rule stage2:
input: "stage1/{id}.1"
output: "stage2/{id}.2"
threads: 2
resources: mem_mb=1024, runtime=5
shell: "cp {input} {output}"
rule stage3:
input: "stage2/{id}.2"
output: "stage3/{id}.3"
threads: 2
resources: mem_mb=2048, runtime=5
shell: "cp {input} {output}"
A typical run of this pipeline might use the command below. Note that for this trivial example we are running from an sinteractive session. However, normally this would be submitted as a batch job.
[user@cn3144]$ snakemake -j5 --latency-wait=180 \
--cluster="sbatch -c {threads} --mem={resources.mem_mb} --time={resources.runtime} -p quick"
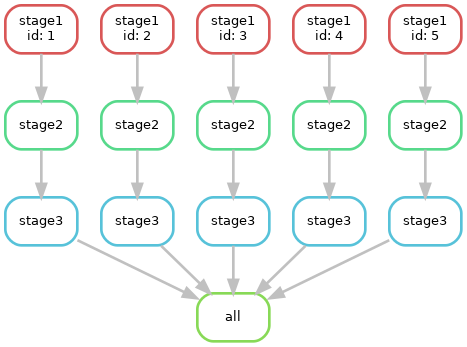
This pipeline ran 15 slurm jobs each with a time limit of 5 minutes, 2 CPUs, and either 1024MB or 2048MB of memory. Obviously each job had a very short actual runtime. Given that it takes a fixed overhead to schedule jobs this is not optimal for cluster efficiency. In addition the queue wait time for each job slows down the pipeline.
We can improve on this by grouping each of the steps for a sample into a
single job. This is done by assigning rules to groups either with the
group: keyword in the snakefile or from the command line like
this:
[user@cn3144]$ snakemake -j5 --latency-wait=180 \
--groups stage1=grp1 stage2=grp1 stage3=grp1 \
--cluster="sbatch -c {threads} --mem={resources.mem_mb} --time={resources.runtime} -p quick"
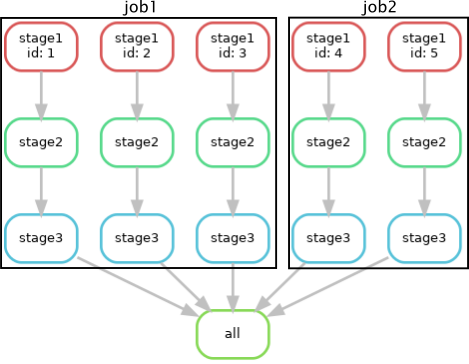
In this example, the number of slurm jobs was reduced from 15 to 5. Each job still used 2 CPUs but was allocated 2048MB of memory (the max). Unfortunately snakemake is not yet smart enough to sum the time instead of taking the max. This may be fixed in the future. Note that it may not make sense to group jobs with very different resource requirements. This is an illustration for what snakemake did:
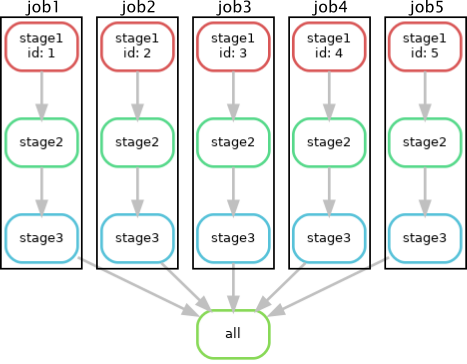
Next we can combine multiple "samples" into a single job. For example, to combine 3 samples per job we could run
[user@cn3144]$ snakemake -j5 --latency-wait=180 \
--groups stage1=grp1 stage2=grp1 stage3=grp1 \
--group-components grp1=3 \
--cluster="sbatch -c {threads} --mem={resources.mem_mb} --time={resources.runtime} -p quick"
This results in 2 jobs - one with 6 CPUs, 6GB, and 15 minute time limit for 3 of the samples and one with 4 CPUs, 4GB, and 10 minutes time limit. Notice how again snakemake is not yet sophisticated enough to distinguish between the requirements for CPUs/memory vs runtime. That may mean that some manual intervention may be needed to make this work properly. Here is the visualization of this run: