Trinity represents a novel method for the efficient and robust de novo reconstruction of transcriptomes from RNA-seq data. Trinity combines three independent software modules: Inchworm, Chrysalis, and Butterfly, applied sequentially to process large volumes of RNA-seq reads. Trinity partitions the sequence data into many individual de Bruijn graphs, each representing the transcriptional complexity at at a given gene or locus, and then processes each graph independently to extract full-length splicing isoforms and to tease apart transcripts derived from paralogous genes. Briefly, the process works like so:
Trinity was developed at the Broad Institute & the Hebrew University of Jerusalem.
In addition to these core functions, Trinity also incudes scripts to do in silico normalization, transcript quantitation, differential expression, and other downstream analyses.
Trinotate, the comprehensive annotation suite designed for automatic functional annotation of transcriptomes, particularly de novo assembled transcriptomes, from model or non-model organisms, is also available.
Trinity creates a lot of temporary files and for efficiency we highly recomment that it be run from lscratch as shown in the examples below. This is especially true for swarms of trinity runs which can result in severe stress on the shared file systems if lscratch is not used. Expect runtime reductions of approximately 10% when using lscratch.
Allocate an interactive session and run the program. Sample session:
[user@biowulf]$ sinteractive --cpus-per-task=6 --gres=lscratch:150 --mem=20g
salloc.exe: Pending job allocation 46116226
salloc.exe: job 46116226 queued and waiting for resources
salloc.exe: job 46116226 has been allocated resources
salloc.exe: Granted job allocation 46116226
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn3144 are ready for job
[user@cn3144]$ module load trinity
[user@cn3144]$ cd /lscratch/$SLURM_JOB_ID
[user@cn3144]$ cp -pr $TRINITY_TEST_DATA/test_Trinity_Assembly .
[user@cn3144]$ cd test_Trinity_Assembly
[user@cn3144]$ Trinity --seqType fq --max_memory 2G \
--left reads.left.fq.gz \
--right reads.right.fq.gz \
--SS_lib_type RF \
--CPU $((SLURM_CPUS_PER_TASK - 2))
[...snip...]
[user@cn3144]$ exit
salloc.exe: Relinquishing job allocation 46116226
[user@biowulf]$
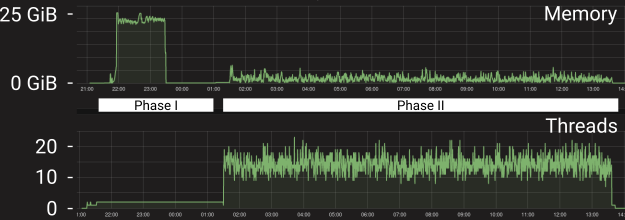
A Trinity run can be devided into two phases - (I) one phase that uses more memory but is not easily parallelized and (II) a phase that uses less memory but can be parallelized. Below is a trace of the memory usage and the number of active threads for a genome based Trinity (2.4.0) assembly of ~15M normalized paired end human reads running on a single node with 10 allocated CPUs:
While Trinity can parallelize Phase II across multiple nodes, we currently do not support this capability on biowulf because it results in too many short jobs. Note that this phase often ends up mildly overloaded with wildly varying CPU load spikes. We have not found a way to properly control this and suggest allocating 2 additional CPUs.
To run Trinity on a single node, create a batch script similar to the following example.
#! /bin/bash
# this file is trinity.sh
function die() {
echo "$@" >&2
exit 1
}
module load trinity/2.14.0 || die "Could not load trinity module"
[[ -d /lscratch/$SLURM_JOB_ID ]] || die "no lscratch allocated"
inbam=$1
mkdir /lscratch/$SLURM_JOB_ID/in
mkdir /lscratch/$SLURM_JOB_ID/out
cp $inbam /lscratch/$SLURM_JOB_ID/in
bam=/lscratch/$SLURM_JOB_ID/in/$(basename $inbam)
out=/lscratch/$SLURM_JOB_ID/out
Trinity --genome_guided_bam $bam \
--SS_lib_type RF \
--output $out \
--genome_guided_max_intron 10000 \
--max_memory 28G \
--CPU 12
mv $out/Trinity-GG.fasta /data/$USER/trinity_out/$(basename $inbam .bam)-Trinity-GG.fasta
Submit this job using the Slurm sbatch command.
biowulf$ sbatch --mem=30g --cpus-per-task=12 --gres=lscratch:150 trinity.sh /data/$USER/trinity_in/sample.bam