UNAFold (Unified Nucleic Acid Folding) is a comprehensive software package for nucleic acid folding and hybridization prediction.
See the UNAFold reference page on the mfold site for a detailed list of references.
Man pages for the commands are available as well.
mfold can process a single sequence in genbank, embl, or fasta format. As an example, let's analyze a yeast tRNA-phe (using a simple constraint file with forces base pairing between the first 7 NTs and their complementary bases from the known structure):
helix$ module load unafold helix$ UNAFold.pl \ -c /usr/local/apps/mfold/TEST_DATA/Phe-GAA-1-1-nointron.constraint \ /usr/local/apps/unafold/TEST_DATA/Phe-GAA-1-1-nointron.fa Checking for boxplot_ng... found, supports Postscript Checking for hybrid-plot-ng... found, supports Postscript, GIF, JPEG, PNG Checking for sir_graph_ng or sir_graph... found, supports Postscript Checking for ps2pdfwr... found Calculating for Phe-GAA-1-1, t = 37 Energy dot plot created. Rotation angle: 0.00 degrees Input File: Phe-GAA-1-1-nointron.fa_1.ct Sequence length: 73 Using 356.0 degrees of the circle Output: Phe-GAA-1-1-nointron.fa_1.ps [...snip...]
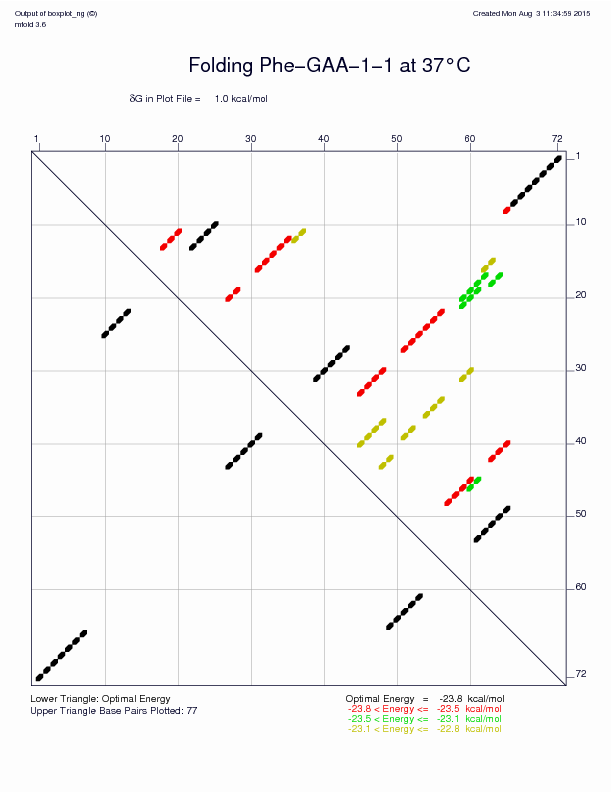
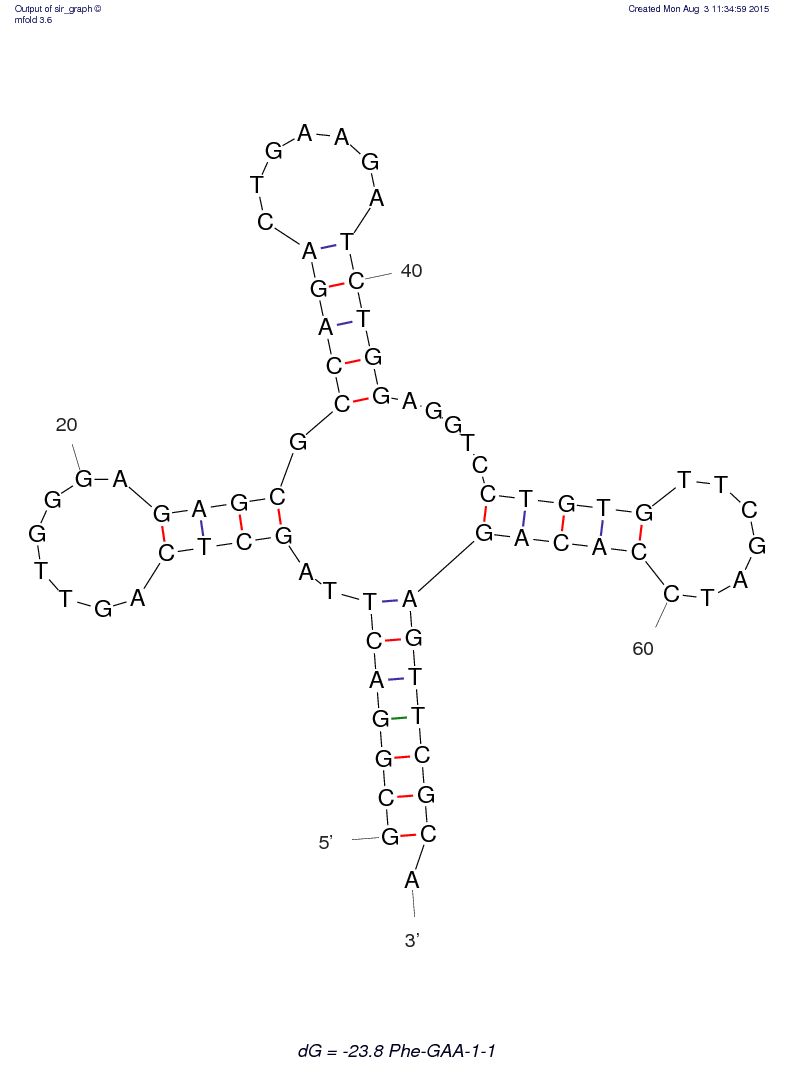
UNAFold will generate output files in different formats for the energy dot plot, the optimal structure, and suboptimal structures (if any). The command above, for example, produces these two graphs amongst others:
|
Energy dot plot |
|
Optimal structure |
The loop containing the 'GAA' anticodon is at the top.
The following batch script will analyze the first 10 sequences in the fasta file, one at a time:
#! /bin/bash
# filename: unafold_batch.sh
set -e
module load emboss || exit 1
module load mfold || exit 2
mkdir -p mfold.out
cd mfold.out
MFA=/usr/local/apps/mfold/TEST_DATA/sacCer1-tRNAs.fa
for sname in $(grep '>' $MFA | grep -v '\?' | awk 'NR < 11 {print substr($1, 2)}'); do
echo "processing $sname"
mkdir -p $sname
cd $sname
seqret -outseq ${sname}.fa ${MFA}:${sname}
UNAFold.pl ${sname}.fa
cd ..
done
A batch process is then started with
biowulf$ sbatch unafold_batch.sh 612486
To process a number of sequences in parallel, set up a swarmfile like this:
UNAFold.pl seq1.fa NA=RNA UNAFold.pl seq2.fa NA=RNA UNAFold.pl seq3.fa NA=RNA
and start your swarm with
biowulf$ swarm -f swarmfile --module unafold 613208
To use mfold interactively, allocated an interactive session with
biowulf$ sinteractive salloc.exe: Granted job allocation 614363 salloc.exe: Waiting for resource configuration salloc.exe: Nodes cn0016 are ready for job cn0016$ UNAFold.pl seq1.fa [...] cn0016$ exit biowulf$
Man pages for the commands are available as well.