IMOD is a set of image processing, modeling and display programs used for tomographic reconstruction and for 3D reconstruction of EM serial sections and optical sections. The package contains tools for assembling and aligning data within multiple types and sizes of image stacks, viewing 3-D data from any orientation, and modeling and display of the image files. IMOD was developed primarily by David Mastronarde, Rick Gaudette, Sue Held, Jim Kremer, Quanren Xiong, and John Heumann at the University of Colorado.
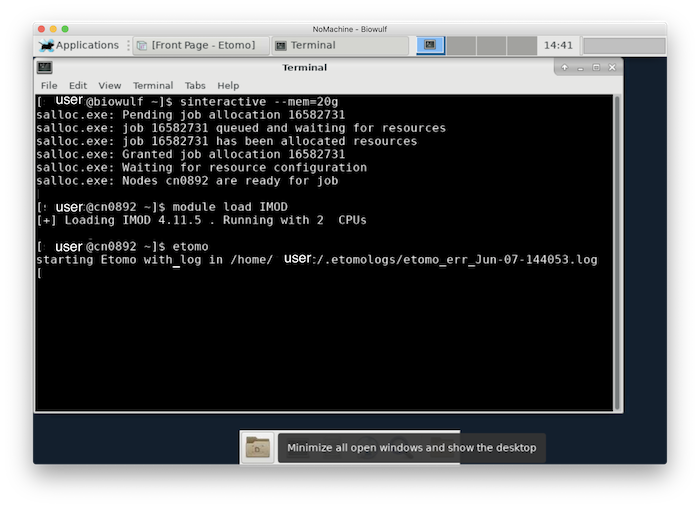
Allocate an interactive session and run the program. Sample session (user input in bold):
[user@biowulf]$ sinteractive --cpus-per-task=4 --mem=10g
salloc.exe: Pending job allocation 46116226
salloc.exe: job 46116226 queued and waiting for resources
salloc.exe: job 46116226 has been allocated resources
salloc.exe: Granted job allocation 46116226
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn3144 are ready for job
[user@cn3144]$ module load IMOD
[+] Loading IMOD 4.11.5 . Running with 4 CPUs
# note: loading the module will cause the environment variable IMOD_PROCESSORS to be set to the number of
# allocated CPUs. i.e. 4 in this example.
[user@cn3144]$ tar xvzf /usr/local/apps/IMOD/imod_data.tar.gz
[user@cn3144]$ mrc2tif imod_data/golgi.mrc imod_data/golgi.tif
Writing TIFF images. ................................
[user@cn3144]$ newstack golgi.mrc golgi.st
RO image file on unit 1 : golgi.mrc Size= 2049 K
This is a byte-swapped file.
This file has an old-style MRC header.
Number of columns, rows, sections ..... 256 256 32
Map mode .............................. 0 (byte)
Start cols, rows, sects, grid x,y,z ... 0 0 0 256 256 32
Pixel spacing (Angstroms).............. 1.000 1.000 1.000
Cell angles ........................... 90.000 90.000 90.000
Fast, medium, slow axes ............... X Y Z
Origin on x,y,z ....................... 0.000 0.000 0.000
Minimum density ....................... 17.000
Maximum density ....................... 195.00
Mean density .......................... 83.733
tilt angles (original,current) ........ 0.0 0.0 0.0 0.0 0.0 0.0
Space group,# extra bytes,idtype,lens . 0 0 0 0
[...]
[user@cn3144 ~]$ exit
salloc.exe: Relinquishing job allocation 46116226
[user@biowulf ~]$
To run etomo or other IMOD graphics applications, you need a graphics connection to Biowulf. We recommend HPCOnDemand. for Windows, Mac or Linux.
Once you have an HPCOnDemand connection to Biowulf, start an interactive session, load the IMOD module, and then run etomo. Sample session following the etomoTutorial.

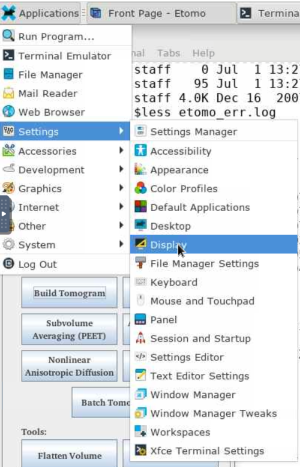
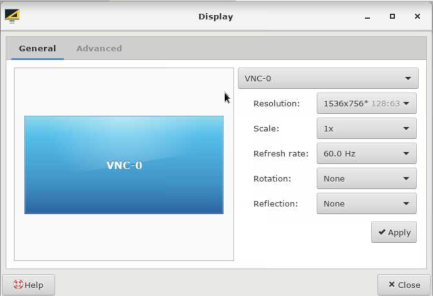
If you are having trouble seeing the entire etomo window, here is an HPCOnDemand tip: Click "Settings" form "Applications" on the top-left corner and then click "Display". And then adjust "Resolution" accordingly.
You should then be able to work through the entire tutorial.
Create a batch input file (e.g. IMOD.sh). For example:
#!/bin/bash set -e module load IMOD cd /data/$USER/myimagedir tif2mrc cell*.tif cell.mrc newstack cell*.mrc cell.st
Submit this job using the Slurm sbatch command.
sbatch [--cpus-per-task=#] [--mem=#] IMOD.sh
Create a swarmfile (e.g. IMOD.swarm). For example:
newstack cell*.mrc newstack cell2*.mrc newstack cell3*.mrc
Submit this job using the swarm command.
swarm -f IMOD.swarm [-g #] [-t #] --module IMODwhere
| -g # | Number of Gigabytes of memory required for each process (1 line in the swarm command file) |
| -t # | Number of threads/CPUs required for each process (1 line in the swarm command file). |
| --module IMOD | Loads the IMOD module for each subjob in the swarm |