Swarm is a script designed to simplify submitting a group of commands to
the Biowulf cluster. Some programs do not scale well or can't use distributed memory.
Other programs may be 'embarrassingly parallel', in that many independent jobs need to be
run. These programs are well suited to running 'swarms of jobs'.
The swarm script simplifies these computational problems.
Note that swarm is NOT a workflow manager. It is merely a convenience
wrapper for the Slurm sbatch --array command.
Swarm reads a list of command lines (termed "commands" or "processes") from a swarm command file (termed the "swarmfile"), then automatically
submits those commands to the batch system to execute.
Command lines in the swarmfile should appear just as they would be entered on a Linux command line.
Swarm encapsulates each command line in a single temporary command script, then submits all command scripts to the Biowulf
cluster as a Slurm job array.
By default, swarm runs one command per core on a node. Thus, a node with 16 cores can run 16 commands in parallel with 16 subjobs.
For example, create a file that looks something like this (NOTE: lines that begin with a #
character are interpreted as comments and are not executed):
[biowulf]$ cat file.swarm
# My first swarmfile -- this file is file.swarm
uptime
uptime
uptime
uptime
Then submit to the batch system:
[biowulf]$ swarm --verbose 1 file.swarm
4 commands run in 4 subjobs, each command requiring 1.5 gb and 1 thread
12345
This will result in a single job (jobid 12345) of four subjobs (subjobids 0, 1, 2, 3), with each swarmfile line being run independently as a single subjob.
By default, each subjob is allocated a 1.5 gb of memory and 1 core (consisting of 2 cpus).
The subjobs will be executed within the same directory from which the swarm was submitted.
The following diagram visualizes how the job array will look:
All output will be written to that same directory. By default, swarm will create two output files for each independent subjob, one for
STDOUT and one for STDERR. The format is name_jobid_subjobid.{e,o}:
Usage: swarm [swarm options] [sbatch options] swarmfile
Basic options:
-g,--gb-per-process [float]
gb per process (default = 1.5)
-t,--threads-per-process [int]
threads per process (default = 1)
-p,--processes-per-subjob,--parallel [int]
processes per subjob (default = 1)
-b,--bundle [int] bundle more than one command line per subjob and
run sequentially (this automatically multiplies the
time needed per subjob)
--noht don't use hyperthreading, equivalent to slurm
option --threads-per-core=1
--usecsh use tcsh as the shell instead of bash
--err-exit exit the subjob immediately on first non-zero exit
status. NOTE: this may fail if the exit status is generated
within a subshell or a forked process.
-m,--module [str] provide a list of environment modules to load prior
to execution (comma delimited)
--no-comment don't ignore text following comment character #
--comment-char [char] use something other than # as the comment character
--maxrunning [int] limit the number of simultaenously running subjobs
--merge-output combine STDOUT and STDERR into a single file per
subjob (.o)
--logdir [dir] directory to which .o and .e files are to be
written (default is current working directory)
--noout completely throw away STDOUT
--noerr completely throw away STDERR
--time-per-command [str] time per command (same as --time)
--time-per-subjob [str] time per subjob, regardless of -b or -p
Development options:
--no-scripts don't create temporary swarm scripts (with --debug
or --devel)
--no-run,--debug don't actually run
--devel combine --debug and --no-scripts, and be very
chatty
-v,--verbose [int] can range from 0 to 6, with 6 the most verbose
--silent don't give any feedback, just jobid
-h,--help print this help message
-V,--version print version and exit
Advanced options:
--prolog [str] command(s) to run per subjob, before swarmfile commands
--prolog-source [str] source prolog file, rather than execute
--prolog-time [str] time needed to run prolog (default 10 min)
--epilog [str] command(s) to run per subjob, after swarmfile commands
--epilog-time [str] time needed to run epilog (default 10 min)
--oldpack do not condense packed output
--joblog create joblog file (with -p)
sbatch options:
-J,--job-name [str] set the name of the job
--dependency [str] set up dependency (i.e. run swarm before or after)
--time [str] change the walltime for each subjob (default is
04:00:00, or 4 hours)
-L,--licenses [str] obtain software licenses (e.g. --licenses=matlab)
--partition [str] change the partition (default is norm)
--gres [str] set generic resources for swarm
--qos [str] set quality of service for swarm
--reservation [str] select a slurm reservation
--sbatch [str] add sbatch-specific options to swarm; these options
will be added last, which means that swarm options
for allocation of cpus and memory take precedence
Environment variables:
The following environment variables will affect how sbatch allocates
resources:
SBATCH_JOB_NAME Same as --job-name
SBATCH_TIMELIMIT Same as --time
SBATCH_PARTITION Same as --partition
SBATCH_QOS Same as --qos
SBATCH_RESERVATION Same as --reservation
The following environment variables are set within a swarm:
SLURM_ARRAY_JOB_ID single slurm jobid for entire swarm
SLURM_JOB_ID unique jobid for each subjob in swarm
SWARM_ARRAY_TASK_ID local numeric id of swarm subjob
SWARM_PROC_ID processor slot on which command is run (-p only)
SWARM_COMM_ID command number from swarmfile
For more information, type "man swarm".
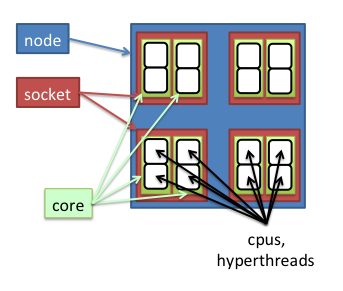
A socket is a receptacle on the motherboard for one physically packaged processor, each can contain one or more cores.
A core is a complete private set of registers, execution units, and retirement queues needed to execute programs.
Nodes on the biowulf cluster can have 8, 16, or 32 cores.
A cpu has the attributes of one core, but is managed and scheduled as a single logical processor by the operating system.
Hyperthreading is the implementation of multiple cpus on a single core.
All nodes on the biowulf cluster have hyperthreading enabled, with 2 cpus per core.
Slurm allocates on the basis of cores. The smallest subjob runs on a single core, meaning the smallest number of cpus that swarm can allocate is 2.
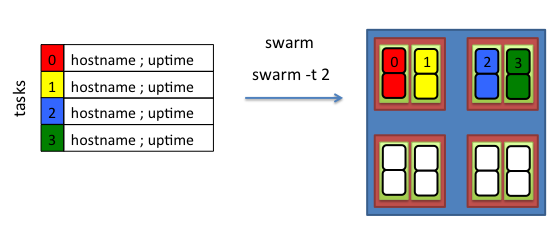
Swarm reads a swarmfile and creates a single subjob per line. By default a subjob is allocated to a single core.
Each line from a swarmfile has access to 2 cpus.
Running swarm with the option -t 2 is thus no different than running swarm without the -t option, as both cpus (hyperthreads)
are available to each subjob.
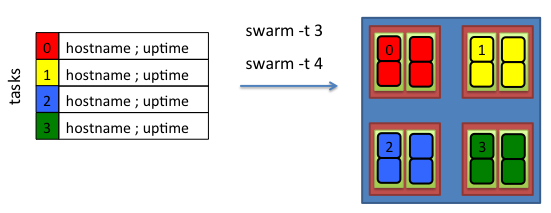
If commands in the swarmfile are multi-threaded, passing the -t option guarantees enough cpus will be available to the generated slurm subjobs.
For example, if the commands require either 3 or 4 threads, giving the -t 3 or -t 4 option allocates 2 cores per subjob.
The nodes on the biowulf cluster are configured to constrain threads within the cores the subjob is allocated. Thus, if a multi-threaded
command exceeds the cpus available, the command will run much slower than normal!
This may not be reflected in the overall cpu load for the node.
Memory is allocated per subjob by swarm, and is strictly enforced by slurm.
If a single subjob exceeds its memory allocation (by default 1.5 GB per swarmfile line), then
the subjob will be killed by the batch system.
See below for examples on how to allocate threads and memory.
More than one swarmfile line can be run per subjob in parallel using the -p option. See below for more information on -p.
If you are unsure of what to expect with a combination of swarm options, swarm can be run in development mode by including
the option --devel. This will display a consise explanation of what swarm will do when submitted.
will create a swarm of 4 subjobs, with each subjob running the single command "uptime".
Bundling
There are occasions when running a single swarmfile line per subjob is inappropriate, such as when commands
are very short (e.g. a few seconds) or when there are many thousands or millions of commands in a swarmfile. In
these circumstances, it makes more sense to bundle the swarm. For example, a swarmfile of 10,000
commands when run with a bundle value of 40 will generate 250 subjobs (10000/40 = 250):
[biowulf]$ swarm --devel -b 40 file.swarm
10000 commands run in 250 subjobs, each requiring 1 gb and 1 thread, running 40 commands serially per subjob
NOTE: If a swarmfile results in more than 1000 subjobs, swarm will automatically autobundle the commands.
ALSO: The time needed per subjob will be automatically multiplied by the bundle factor. If the total time
per subjob exceeds the maximum walltime of the partition, an error will be given and the swarm will not be submitted.
Comments
By default, any text on a single line that follows a # character is assumed to be a comment,
and is ignored. For example,
[biowulf]$ cat file.swarm
# Here are my commands
uptime # this gives the current load status
pwd # this gives the current working directory
hostname # this gives the host name
However, there are some applications that require a # character in the input:
The option --no-comment can be given to avoid removal of text following the # character.
Alternatively, another comment character can be designated using the --comment-char option.
Command lists
Multiple commands can be run serially (one after the other) when they are separated by a semi-colon (;). This
is also known as a command list. For example,
[biowulf]$ cat file.swarm
hostname ; date ; sleep 200 ; uptime
hostname ; date ; sleep 200 ; uptime
hostname ; date ; sleep 200 ; uptime
hostname ; date ; sleep 200 ; uptime
[biowulf]$ swarm file.swarm
will create 4 subjobs, each running independently on a single cpu. Each subjob will run "hostname", followed
by "date", then "sleep 200", then "uptime", all in order.
Complex commands
Environment variables can be set, directory locations can be changed, subshells can be spawned all within
a single command list, and conditional statements can be given. For example, if you wanted to run some
commands in a newly created random temporary directory, you could use this:
[biowulf]$ cat file.swarm
export d=/data/user/${RANDOM} ; mkdir -p $d ; if [[ -d $d ]] ; then cd $d && pwd ; else echo "FAIL" >&2 ; fi
export d=/data/user/${RANDOM} ; mkdir -p $d ; if [[ -d $d ]] ; then cd $d && pwd ; else echo "FAIL" >&2 ; fi
export d=/data/user/${RANDOM} ; mkdir -p $d ; if [[ -d $d ]] ; then cd $d && pwd ; else echo "FAIL" >&2 ; fi
export d=/data/user/${RANDOM} ; mkdir -p $d ; if [[ -d $d ]] ; then cd $d && pwd ; else echo "FAIL" >&2 ; fi
NOTE: By default, command lists are interpreted as bash commands. If a swarmfile contains tcsh- or csh-specific
commands, swarm may fail unless --usecsh is included.
Line continuation markers
Application commands can be very long, with dozens of options and flags, and multiple commands separated by
semi-colons. To ease file editing, line continuation markers can be used to break up the single swarm commands
into multiple lines. For example, the swarmfile
All swarm options can be incorporated into the swarmfile using swarmfile directives. Options preceded by #SWARM in the swarmfile (flush against the left side) will be evaluated the same as command line options.
For example, if the contents of swarmfile is as follows:
then each subjob will request 4 cpus, 10 GB of RAM and 10 minutes of walltime. The amount of memory and walltime requested with command line options and the partition chosen with the SBATCH_PARTITION environment variable supersedes the amount requested with swarmfile directives.
NOTE: All lines with correctly formatted #SWARM directives will be removed even if --no-comment or a non-default --comment-char is given.
STDOUT and STDERR output from subjobs executed under swarm will be
directed to a file named swarm_jobid_subjobid.o and swarm_jobid_subjobid.e, respectively.
Please pay attention to the memory requirements of your swarm jobs!
When a swarm job runs out of memory, the node stalls and the job is eventually killed or
dies.
At the bottom of the .e file, you may see a warning like this:
slurmstepd: Exceeded job memory limit at some point. Job may have been partially swapped out to disk.
If a job dies before it is finished, this output may not be available. Contact
staff@hpc.nih.gov when you have a question about why
a swarm stopped prematurely.
Renaming output files
The sbatch option --job-name can be used to rename the default output files.
Combining STDOUT and STDERR into a single file per subjob
Including the --merge-output option will cause the STDERR output to be combined into the file used
for STDOUT. For swarm, that means the content of the .e files are written to the .o file. Keep in mind that
interweaving of content will occur.
By default, the STDOUT and STDERR files are written to the same directory from which the swarm
was submitted. To redirect the files to a different directory, use --logdir:
Be aware of programs that write directly to a file using a fixed filename.
A file will be overwritten and garbled if multiple processes are writing to the same file.
If you run multiple instances of such programs then for each instance you will
need to a) change the name of the file in each command or b) alter the path to the file. See
the EXAMPLES section for some ideas.
To see how swarm works, first create a file containing a few simple
commands, then use swarm to submit them to the batch queue:
[biowulf]$ cat > file.swarm
date
hostname
ls -l
^D
[biowulf]$ swarm file.swarm
Use sjobs to monitor the status of your request; an
"R" in the "St"atus column indicates your job is running.
This particular example will probably run to completion
before you can give the qstat command. To see the output from the commands, see
the files named swarm_#_#.o.
By default any swarmfile with > 1000 commands will be autobundled
unless it is deliberately bundled with the -b flag.
If you have over 1000 commands, especially if each one runs for a short
time, you should 'bundle' your jobs with the -b flag. For example, if the
swarmfile contains 2560 commands, the following swarm command will group them into
bundles of 40 commands each, producing 64 command bundles. Swarm will then submit the
64 command bundles, rather than the 2560 commands individually, as a single swarm job.
This would result in a swarm of 64 (2560/40) subjobs.
[biowulf]$ swarm -b 40 file.swarm
Note that commands in a bundle will run sequentially on the assigned node.
Allocating memory and threads with -g and -t options
If the subjobs require significant amounts of memory (> 1.5 GB) or threads (> 1 per core), a swarm can
run fewer subjobs per node than the number of cores available
on a node. For example, if the commands in a swarmfile need up to 40 GB of
memory each using 8 threads, running swarm with --devel shows what might happen:
[biowulf]$ swarm -g 40 -t 8 --devel file.swarm
14 commands run in 14 subjobs, each requiring 40 gb and 8 threads
The default partition norm has nodes with a maximum of 248GB memory. If -g exceeds 373GB, swarm will give a warning message:
The sbatch options --exclusive and --mem=0 are not allowed with swarm. The use of these options very often wastes resources. Instead, users should test their commands and determine the optimum number of threads or amount of memory to use per command. If these options are required, you will need to submit a batch script using sbatch.
Using -p option to "pack" or "parallelize" commands
By default, swarm allocates a single command line per subjob. If the command is single-threaded, then swarm wastes half the
cpus allocated, because the slurm batch system allocates no less than a single core (or two cpus) per subjob. It is also sometimes more efficient
to run multiple commands in parallel on the same node with a single allocation of /lscratch.
In order to use all the cpus allocated to a swarm, the option -p will set the number of commands run per subjob. The value of -p
can be any even number, and corresponds to the number of cpus allocated per subjob. For example, running 64 commands
in the swarmfile using -p 16 allocates 16 cpus per subjob, one subjob per node. Each subjob runs up to 16 processes simultaneously:
[biowulf]$ swarm -p 16 --time 60 work_64.swarm
1001
...
[biowulf]$ sjobs -u user
User JobId JobName Part St Runtime Walltime Nodes CPUs Memory Dependency Nodelist
========================================================================================================================
user 1001_0 swarm norm R 20:42 60:00 1 16 24 GB cn0048
user 1001_1 swarm norm R 20:42 60:00 1 16 24 GB cn0054
user 1001_2 swarm norm R 20:42 60:00 1 16 24 GB cn4278
user 1001_3 swarm norm R 20:42 60:00 1 16 24 GB cn4303
========================================================================================================================
cpus queued = 0
cpus running = 48 / 64
mem queued = 0.0 B
mem running = 56.6 GB / 97.5 GB
jobs queued = 0
jobs running = 4
While the subjobs and processes are running, each subjob/node gets outputs its own STDOUT and STDERR to .o and .e files (in this case _0, _1, _2, ... _15). Additionally, there is a global .o and .e file labeled
with "_p".
Normally when the subjobs complete, the .o and .e files are consolidated into single files per subjob and individual output
is segmented in each subjob output:
The contents of the joblog files consists of six columns: unique_jobid, command number, start time, end time, elapsed time (in seconds), and exit code of the command.
In the case where each swarm subjob must create or use a unique directory or file, an environment variable SWARM_PROC_ID is
available to discriminate the processes running with -p.
For example, in order to create a unique directory in allocated /lscratch for each subjob, this bash code example can be used:
In this case, while the files created within each distinct /lscratch/${SLURM_JOB_ID}/${SLURM_ARRAY_JOB_ID}_${SLURM_ARRAY_TASK_ID}_${SWARM_PROC_ID} directory are identical to all
the other swarm subjobs, the final tarball is unique:
By default all jobs and subjobs have a walltime of 2 hours. If a swarm subjob exceeds its walltime, it will be killed!.
On the other hand, if your swarm subjobs have a very short walltime, then their priority on the queue may be elevated. Therefore,
it is best practice to set a walltime using the --time option that reflects the estimated execution time of the subjobs.
For example, if the command lines in a swarm are expected to require no more than half an hour to complete, the swarm command should be:
[biowulf]$ swarm --time 00:30:00 file.swarm
Because a subjob is expected to be running a single command from the swarmfile, the value of --time can be considered
the amount of time to run a single command. When a swarm is bundled, the value for --time is then
multiplied by the bundle factor. For example, if
a swarm that normally creates 64 commands is bundled to run 4 commands serially, the value of --time is
multiplied by 4:
[biowulf]$ swarm --time 00:30:00 -b 4 --devel file.swarm
64 commands run in 16 subjobs, each command requiring 1.5 gb and 1 thread, running 4 processes serially per subjob
sbatch --array=0-15 --job-name=swarm --time=2:00:00 --cpus-per-task=2 --partition=norm --mem=1536
If a swarm has more than 1000 commands and is autobundled, there is a chance that the time requested will exceed
the maximum allowed. In that case, an error will be thrown:
ERROR: Total time for bundled commands is greater than partition walltime limit.
Try lowering the time per command (--time=04:00:00), lowering the bundle factor
(if not autobundled), picking another partition, or splitting up the swarmfile.
There are two additional options for setting the time of a swarm. --time-per-command is identical to --time, and
merely serves as a more obvious explanation of time allocation.
--time-per-subjob overrides the time adjustments applied when bundling or packing commands.
This option can be used when a single command takes less than 1 minute to complete and there are a high number of commands bundled per
subjob:
[biowulf]$ swarm --time-per-subjob 00:30:00 -b 4 --devel file.swarm
64 commands run in 16 subjobs, each command requiring 1.5 gb and 1 thread, running 4 processes serially per subjob
sbatch --array=0-15 --job-name=swarm --time=30:00 --cpus-per-task=2 --partition=norm --mem=1536
If a swarm is run as a single step in a pipeline, job dependencies can be handled with the --dependency options.
For example, a first script (first.sh) is to be run to generate some initial data files. Once this job is finished, a swarm of
commands (swarmfile.txt) is run to take the output of the first script and process it. Then, a last script (last.sh) is run
to consolidate the output of the swarm and further process it into its final form.
Below, the swarm is run with a dependency on the first script. Then the last script is run with a dependency on the swarm.
The swarm will sit in a pending state until the first job (10001) is completed, and the last job will sit in a pending state until
the entire swarm (10002) is completed.
The jobid of a job can be captured from the sbatch command and passed to subsequent submissions in a script (master.sh).
For example, here is a bash script which automates the above procedure, passing the variable $id to the first script. In this way,
the master script can be reused for different inputs:
[biowulf]$ squeue -u user
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
10002_[0-3] norm swarm user PD 0:00 1 (Dependency)
10003 norm last.sh user PD 0:00 1 (Dependency)
10001 norm first.sh uwer R 0:33 1 cn0121
The dependency key 'afterany' means run only after the entire job finishes, regardless of its exit status. Swarm passes the exit
status of the last command executed back to Slurm, and Slurm consolidates all the exit statuses of the subjobs in the job array into
a single exit status.
The final statuses for the jobs can be seen with sacct. The individual subjobs from swarm are designated
by jobid_subjobid:
[biowulf]$ sacct
JobID JobName Partition Account AllocCPUS State ExitCode
------------ ---------- ---------- ---------- ---------- ---------- --------
10001 first.sh norm user 2 COMPLETED 0:0
10001.batch batch user 1 COMPLETED 0:0
10002_3 swarm norm user 2 FAILED 2:0
10002_3.bat+ batch user 1 FAILED 2:0
10003 last.sh norm user 2 COMPLETED 0:0
10003.batch batch user 1 COMPLETED 0:0
10002_0 swarm norm user 2 COMPLETED 0:0
10002_0.bat+ batch user 1 COMPLETED 0:0
10002_1 swarm norm user 2 COMPLETED 0:0
10002_1.bat+ batch user 1 COMPLETED 0:0
10002_2 swarm norm user 2 COMPLETED 0:0
10002_2.bat+ batch user 1 COMPLETED 0:0
If any of the subjobs in the swarm failed, the job is marked as FAILED. It almost all cases, it is better
to rely on afterany rather than afterok, since the latter may cause the dependent job to
remain queued forever:
[biowulf]$ sjobs
................Requested............................
User JobId JobName Part St Runtime Nodes CPUs Mem Dependency Features Nodelist
user 10003 last.sh norm PD 0:00 1 1 2.0GB/cpu afterok:10002_* (null) (DependencyNeverSatisfied)
NOTE: Setting -p causes multiple commands to run per subjob. Because of this, the exit status of the
subjob can come from any of the multiple processes in the subjob. Include --joblog when running with -p to output
a file that lists the exit code along with the itemized processes.
If a program writes to a fixed filename, then you may need to run the
program in different directories. First create the necessary directories (for
instance run1, run2), and in the swarmfile cd to the unique output
directory before running the program: (cd using either an absolute path
beginning with "/" or a relative path from your home directory). Lines with
leading "#" are considered comments and ignored.
[biowulf]$ cat > file.swarm
# Run ped program using different directory
# for each run
cd pedsystem/run1; ../ped
cd pedsystem/run2; ../ped
cd pedsystem/run3; ../ped
cd pedsystem/run4; ../ped
...
[biowulf]$ swarm file.swarm
Running mixed asynchronous and serial commands in a swarm
There are occasions when a single swarm command can contain a mixture of asynchronous and serial commands. For
example, collating the results of several commands into a single output and then running another command on the pooled
results. If run interactively, it would look like this:
It would be more efficient if the four cmdA commands could run asynchronously (in parallel), and then
the last cmdB command would wait until they were all done and then run, all on the same node and in the same
swarm command. This can be achieved using process substitution with this one-liner in a swarmfile:
Here, the cmdA commands are all run asynchronously in four background processes, and the wait command
is given to prevent cmdB from running until all the background processes are finished. Note that line
continuation markers were used for easier editing.
It is sometimes difficult to set the environment properly before running commands. The
easiest way to do this on Biowulf is with
environment modules. Running commands via swarm complicates the issue, because the modules
must be loaded prior to every line in the swarmfile. Instead, you can use the --module
option to load a list of modules:
There are occasions where a set of commands should be run once per subjob or per node, either
before or after the input swarmfile commands are run. This can be facilitated using --prolog
and --epilog.
For example, copying a very large file to local scratch prior to running a set of commands, all
of which use that file as a reference. This is advantageous, as it distributes the I/O load to
the nodes and improves I/O performance.
If work is performed and files are written to /lscratch during the job, the --epilog option can be used to move those files after all commands are finished.
--prolog-source
If a set of commands requires being run in the current shell prior to running commands, for example setting environment variables or initiating a specific conda environment, --prolog-source should be used.
Note that both --prolog and --prolog-source can be used together.
Running prolog or epilog commands can take a substantial amount of time. For example, copying several hundred gb of data to /lscratch can take 30-60 minutes. By default, swarm allocates an additional 10 minutes of time to complete the prolog or epilog commands. --prolog-time and --epilog-time can be used to increase (or decrease) the amount of time necessary for these steps.
For example, copying a large amount of data to /lscratch:
Including --gres=lscratch:N, where N is the number of GB required, will create a subdirectory on the node
corresponding to the jobid, e.g.:
/lscratch/987654/
This local scratch directory can be accessed dynamically using the $SLURM_JOB_ID environment variable:
/lscratch/$SLURM_JOB_ID/
/lscratch/$SLURM_JOB_ID is a temporary work directory. Each swarm subjob should do most if not all of its work in this temporary work directory. This means that any input data should be copied the /lscratch before running any commands, and the output should be copied back to the original location after completion.
Here is a generic example of how to use /lscratch in a swarm using --prolog-source:
Local scratch space is allocated per subjob. By default, that means each command or command list (single line in
swarmfile) is allocated its own independent local scratch space. HOWEVER, there are two situations where some
thought must be given to local scratch space:
bundled swarms (-b): Bundled swarms serialize multiple commands into a single subjob. Since local scratch space is
allocated per subjob, this means that each command in the job inherits the same local scratch space. This means that each
command should be written to deal with any "leftover" files from the previous commands. A simple solution might be to
clean out the local scratch space at the end of each command. For example:
cd /lscratch/$SLURM_JOB_ID ; command1 arg1 arg2 ; rm -rf /lscratch/$SLURM_JOB_ID/*
packed swarm (-p): If a packed swarm using the -p option is given to swarm, then allocated local scratch space is shared
between multiple commands in a single subjob. In this case, make sure to allocate enough local scratch space to accomodate the number of parallel processes designated by -p.
By default, a swarm will inherit the current environment and propagate it to the slurm job. This means any environment variables set at the time of swarm submission will be available during the swarm.
[biowulf]$ GLOBAL_VAR=5 swarm file.swarm
If an entire swarm requires one or more environment variables to be set, the sbatch option --export
can be used to set the variables prior to running. In this example, we need to set the BOWTIE_INDEXES environment variable
to the correct path for all subjobs in the swarm:
NOTE: Environment variables inherited from the current shell or set with the --sbatch "--export=" option are defined
PRIOR to the job being submitted. This prevents setting environment variables using Slurm-generated environment
variables, such as $SLURM_JOB_ID or $SLURM_MEM_PER_NODE.
If each command line in the swarm requires a unique set of environment variables (per command), this must be done in the swarmfile. For example, setting TMPDIR to a unique subdirectory of /lscratch/$SLURM_JOB_ID:
Further, if individual commands within each command line require unique environment variables, this can be done by prefacing the command itself with the variable set:
Swarm creates a single job array via the sbatch command
; all valid sbatch commandline options are also valid for swarm.
However, they must be passed with a single --sbatch option, surrounded
by quotation marks. In this example some extra sbatch options are added.
--mail-type=FAIL: causes a single email per swarm to be sent if one subjob fails for some reason
--mail-type=END,ARRAY_TASKS: causes each subjob to issue an email when the subjob ends for any reason
Unless the ARRAY_TASKS option is specified, mail notifications on job BEGIN, END and FAIL apply to a job array as a whole rather than generating individual email messages for each task in the job array. See userguide.html#email for more information about email notifications.
--export=var=100,nctype=12: sets two environment variables var and nctype to 100 and 12 prior to running
--chdir=/data/user/test: relocate to that directory prior to running any commands
NOTE: Sbatch options passed through swarm using the --sbatch option are listed last in the sbatch command, and thus will override swarm
options. When bundling or packing commands, DO NOT use --time, --cpus-per-task, --mem, or --mem-per-cpu sbatch options, as they will
inevitably conflict with those values set by swarm per command.
Before submitting a large complex swarm to the batch system, it is better
to see what would happen before it's too late. In this case, the --devel option
will display a good deal of information. This example shows a huge number of
commands autobundled to run 346 command lines serially per core.
[biowulf]$ swarm --devel file.swarm
345029 commands run in 998 subjobs, each requiring 1 gb and 1 thread, running 346 commands serially per subjob
--verbose accepts a number between 0 (the same as --silent) and 6. Increasing the verbosity level with --verbose and including --devel will give a visual representation
of the swarm, along with lots of information about the swarm:
This shows a swarm of 32 commands (show as "cmd 0" ==> "cmd 31") within 2 subjobs. Each command requires 5 gb of memory,
and the commands are bundled to run 4 commands sequentially on the cpus allocated.
Users will typically want to write a script to create a large swarmfile. This script can be written in any scripting language, such as bash, perl, or the language of your choice. Some examples are given below to get you started.
Example 1: processing all files in a directory
Suppose you have 800 image files in a directory. You want to set up a swarm job to run an FSL command (e.g. 'mcflirt') on each one of these files.
# this file is make-swarmfile
cd /data/user/mydir
touch swarm.cmd
for file in `ls`
do
echo "mcflirt -in $file -out $file.mcf1 -mats -plots -refvol 90 -rmsrel -rmsabs" >> swarm.cmd
done
Execute this file with
bash make-swarmfile
You should get a file called swarm.cmd which is suitable for submission to the swarm command.
Example 2: Use swarm to pull sequences out of the NCBI nt blast database.
Suppose you have a file containing 1,000,000 GI numbers of sequences. You want to pull these sequences out of the Helix/Biowulf NCBI nt Blast database. You can divide your GI file into chunks, and run a swarm of jobs, each one working on one chunk of GIs, to pull these sequences out of the database.
#!/usr/local/bin/perl
$dir = `pwd`;
# create a directory for all the output sequences
mkdir "${dir}/seqs";
chdir "${dir}/seqs";
# split the list of GIs (in file ../gi.list) into files containing 10000 GIs each.
# The 'split' command will create files called xaa, xab etc.
system("split -l 10000 ../gi.list");
# get a list of all these files
my @files = </$dir/seqs/*>;
# create the swarmfile -- you will need to load the blast/2.2.26 module first
open (SWARM, "> swarm.cmd");
foreach $file (@files) {
print SWARM "cd ${dir}/seqs; fastacmd -p F -d /fdb/blastdb/nt -i $file -o $file.fas\n";
}
close (SWARM);
print "Submitting swarm jobs\n";
`swarm -f ${dir}/seqs/swarm.cmd --module blast/2.2.26`;
Once the swarm jobs are complete, you could if desired combine all the sequences into a single file with
Swarm is available for download here.
Keep in mind that swarm was written for our own systems. It will need to
be adapted for other batch systems to work properly.