The continued growth and support of NIH's Biowulf cluster is dependent upon its demonstrable value to the NIH Intramural Research Program. If you publish research that involved significant use of Biowulf, please cite the cluster. Suggested citation text:
This work utilized the computational resources of the NIH HPC Biowulf cluster (https://hpc.nih.gov).
Why a job remains pending depends on a number of factors, roughly in this order of relevance:
There are some simple steps you can do to speed up the scheduling of your job.
# jobhist 46435925
JobId : 46435925
User : $USER
Submitted : 20170801 15:36:28
Started : 20170801 15:36:48
Submission Path : /data/$USER/rna-seq/oligos
Submission Command : sbatch --array=0-459 --output=/data/$USER/rna-
seq/oligos/Oligos2_%A_%a.o --error=/data/$USER/rna-
seq/oligos/Oligos2_%A_%a.e --cpus-per-task=2
--job-name=Oligos2
--mem=4096 --partition=norm --time=6-16:00:00
/spin1/swarm/$USER/46435925/swarm.batch
Swarm Path : /data/$USER/rna-seq/oligos
Swarm Command : /usr/local/bin/swarm -f oligos.swarm1 -g 4 -t 2 --time 160:00:00
This tells you that the swarmfile corresponding to this swarm was /data/$USER/rna-seq/oligos/oligos.swarm1. You can resubmit it by cd'ing to the Submission Path,
and submitting using the Swarm Command from the info above. In this case:
cd /data/$USER/rna-seq/oligos /usr/local/bin/swarm -f oligos.swarm1 -g 4 -t 2 --time 160:00:00If the swarm subjobs were failing due to lack of memory, of course you will want to increase the memory allocation, by increasing the -g # parameter. Likewise, if the failures were due to walltime limits, you would increase the walltime with --time xxxx.
Suppose you submitted a swarm of 1000 commands, and you see (either from your swarm.o/swarm.e files, or from 'jobhist jobnumber') that subjobs 3, 5, 18 failed.
(Note: snakemake makes it easy to rerun only the failed steps in a pipeline. You might consider using snakemake if you have complicated pipelines and frequently need to rerun subjobs. This FAQ entry deals with an occasional such failure.)
# jobhist 46435925
JobId : 46435925
User : $USER
Submitted : 20170801 15:36:28
Started : 20170801 15:36:48
Submission Path : /data/$USER/rna-seq/oligos
Submission Command : sbatch --array=0-459 --output=/data/$USER/rna-
seq/oligos/Oligos2_%A_%a.o --error=/data/$USER/rna-
seq/oligos/Oligos2_%A_%a.e --cpus-per-task=2
--job-name=Oligos2
--mem=4096 --partition=norm --time=6-16:00:00
/spin1/swarm/$USER/46435925/swarm.batch
Swarm Path : /data/$USER/rna-seq/oligos
Swarm Command : /usr/local/bin/swarm -f oligos.swarm1 -g 4 -t 2 --time 160:00:00
Jobid Partition State Nodes CPUs Walltime Runtime MemReq MemUsed Nodelist
46435925_0 norm COMPLETED 1 2 6-16:00:00 1-01:38:35 4.0GB/node 3.0GB cn3415
46435925_1 norm COMPLETED 1 2 6-16:00:00 3-14:18:34 4.0GB/node 3.0GB cn3444
46435925_2 norm COMPLETED 1 2 6-16:00:00 3-14:14:52 4.0GB/node 3.0GB cn3461
46435925_3 norm FAILED 1 2 6-16:00:00 1-01:38:35 4.0GB/node 4.0GB cn3415
[...]
46435925_5 norm FAILED 1 2 6-16:00:00 1-01:38:35 4.0GB/node 4.0GB cn3415
46435925_18 norm FAILED 1 2 6-16:00:00 1-01:38:35 4.0GB/node 4.0GB cn3415
[...]
In this case, it looks like the failed jobs used the full 4 GB allocated memory, so they probably failed due to memory. (You can also check this on the dashboard). Since most of the jobs ran successfully with a 4 GB memory allocation, you probably don't want to resubmit the entire swarm with an increased memory allocation.
Swarm creates its own set of batch scripts, one for each job, in a directory called /spin1/swarm/$USER/jobid and submits those to the batch system. That directory contains a swarm.batch file, and a cmd.0 file corresponding to subjob 0, cmd.1 file corresponding to subjob 1..etc... You can resubmit only the failed jobs by pulling out the swarm-created commands corresponding to the failed jobs, and resubmit those as a new swarm. e.g. for the example above, pull out the commands corresponding to subjobs 3,5,18 into a new swarm file:
% cd /spin1/swarm/$USER/46435925/ % cat cmd.3 cmd.5 cmd.18 > /data/$USER/new_swarmCheck the number of lines in the new_swarm file. (should be 3 lines corresponding to the 3 subjobs)
% wc -l /data/$USER/new_swarm 3Resubmit requesting 8 GB of memory per subjob, and leaving the threads and walltime the same.
% swarm -f /data/$USER/new_swarm -g 8 -t 2 --time 160:00:00
If a job fails, you might want to examine the tmp files in /lscratch/$SLURM_JOBID. In that case, you would want to set up your batch script to check for failure of a command, and if it fails, copy the /lscratch/$SLURM_JOBID directory back to /scratch or your /data area before the job exits. There is no built-in mechanism in sbatch or slurm to set this up. Each user's definition of 'failure' may be different (e.g. a command may complete but the output file is size 0) and each user may have specific actions they want taken if a command in their script fails. The example below is one suggestion of how to set up a batch script that 'rescues' /lscratch/$SLURM_JOBID if a command fails.
This file jobscript.sh was submitted with 'sbatch jobscript.sh'.
#!/bin/bash
# allocate 50 GB of local disk on the allocated node
#SBATCH --gres=lscratch:50
# send email if the job fails
#SBATCH --mail-type=FAIL
cd /lscratch/$SLURM_JOBID
module load samtools
# copy the data from the /data/$USER area to the local disk
cp /data/$USER/HLA-PRG-LA/NA12878.mini.cram .
# run samtools index
samtools index NA12878.mini.cram
# test if file exists and is non-zero size. If not, tar the entire /lscratch/$SLURM_JOBID area to /scratch/$USER
if [ ! -s NA12878.mini.cram.bai ]
then
echo "samtools index failed. "
echo "Tarring and copying /lscratch/$SLURM_JOBID to /scratch/$USER/lscratch_${SLURM_JOBID}.tar.gz"
tar -cvzf /scratch/$USER/lscratch_${SLURM_JOBID}.tar.gz /lscratch/$SLURM_JOBID
exit 1
fi
# if the file exists and is non-zero size, run the next command
module load HLA-PRG-LA/f0833ed
HLA-PRG-LA.pl --BAM NA12878.mini.cram --graph PRG_MHC_GRCh38_withIMGT --sampleID NA12878 --maxThreads $cpus --workingDir .
# copy the final output back to /data/$USER
cp -r NA12878.mini.hla /data/$USER/HLA-PRG-LA/
In the example batch script above, if the samtools index fails and the output file NA12878.mini.cram.bai does not exist, or is a size-zero file, the user will get an email saying the job failed. The slurm output file slurm-#####.out will report that the samtools index failed, and the location of the tar file that is created.
Your laptop or desktop will almost always do a better job displaying graphics than a remote computational resource like Biowulf. Graphics hardware is designed to be connected directly to a monitor.
When you use a graphical application on a remote system like Biowulf (outside of the visual partition), you are still using your local hardware to display graphics. Intensive graphics computations such as 3D rendering will run into bottlenecks because instead of a GPU performing the calculations and presenting the information directly on a local monitor, a CPU will need to package the required information and send it over the network. This means that all graphics performance will be degraded and that 3D graphics applications in particular will run very slowly if they run at all.
On Linux, there are a collection of libraries and applications that can be used to run graphical applications on remote resources (assuming those resources have graphics hardware). You can take advantage of these libraries by using the visual partition which is composed of nodes containing GPU hardware set aside for graphics visualization. Please bear in mind that graphics performance will still be substantially lower on these nodes than the performance you can achieve on your local workstation connected directly to your monitor. This is because of network latency.
There are some common problems that users of grapical applications run into on Biowulf. Some of these can be improved using the following troubleshooting steps, while a few are just indicative of the nature of Biowulf as a remote computational resource.
mv ~/.bashrc ~/.bashrc.ORIG cp -p /etc/skel/.bashrc ~ mv ~/.bash_profile ~/.bash_profile.ORIG cp -p /etc/skel/.bash_profile ~ module purgeNow start a new Biowulf session, load only the modules needed for your graphical application, and test again. If it works without problems, something in your .bashrc (e.g. a conda environment, see below), .bash_profile, or another module is causing the problem. Add any personal modifications back one by one to your .bashrc and .bash_profile until you identify the cause.
If you still have errors or problems with your graphics, then the environment is not the cause. You can return your environment to its former state with:
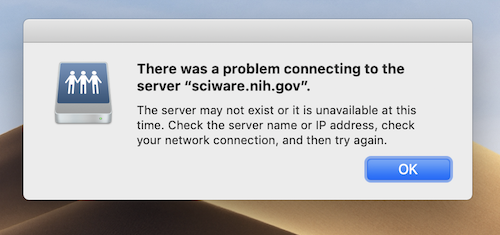
mv ~/.bashrc.ORIG ~/.bashrc mv ~/.bash_profile.ORIG ~/.bash_profileIf you have a non-standard shell (csh, tcsh or zsh), you can follow the same general idea with the corresponding startup files (e.g. .cshrc). If you aren't sure what these files are, please contact staff@hpc.nih.gov
For many users, using Graphical Sessions to display remote graphics will suffice. For those doing complex graphics, benchmarking suggests that the graphics performance obtained from running Matlab directly on your desktop or laptop will be much better than running Matlab remotely on Biowulf compute nodes.
Your /home directory has a quota of 16 GB which cannot be increased and if it fills up a lot of things can go wrong. To find which directory is eating your /home space:
dust $HOMEHere are some reasons your home directory might be full:
~/.cache/pip) can get surprisingly large for frequent pip users.
Quick solution:
rm -rf ~/.cache/pip
~/.singularity/cache,
and can readily be moved to your data directory by setting
export SINGULARITY_CACHEDIR=/data/$USER/.singularitymanually or in your
~/.bashrc.
~/.vep. Make sure to
include --cache --dir_cache $VEP_CACHEDIR with all VEP commands.NOTE: You can also find the number of files per subdirectory by including the -f option to dust:
dust -f $HOME
When contacting the HPC staff with questions, the more relative information you can provide, the better. It will help us to resolve your problem quickly, if we can replicate the problem ourselves or solve it with the information you provided with out asking many additional questions.
Possible shortcut: If there is a generic error message, doing a Google search on that error message will often produce the answer.
The minimum information we need
Examples of excellent problem report:
From: xx.xx@nih.gov To: staff@hpc.nih.gov Subject: problem with XXX version 1.2.0 The module XXX/1.1.0 works, but 1.2.0 gives an error. The error file is in /data/user/mydir [user@biowulf]$ sinteractive [user@cn3133]$ module load XXX/1.1.0 [user@cn3133]$ XXX /A/B/C/YYY 2>XXX_1.1.0.noerror [user@cn3133]$ module load XXX/1.2.0 [user@cn3133]$ XXX /A/B/C/YYY 2>XXX_1.2.0.error
From: xx.xx@nih.gov To: staff@hpc.nih.gov Subject: batch job problem I ran two similar jobs, but one succeeded (job 12345) and one failed (job id 456789). I can't figure out why the failed job failed. The slurm output files are in directory /data/user/mydir. Could you help?
Swarm requires a 'swarm command file' with a command line for each run. In this case, each line would run the desired program against a single file. Here is a sample swarm command file:
blat /fdb/genome/hg19/chr10.fa indir/gi_615762_gb_T33664 -maxGap=3 outdir/gi_615762_gb_T33664.blat blat /fdb/genome/hg19/chr10.fa idir/gi_615763_gb_T33665 -maxGap=3 outdir/gi_615763_gb_T33665.blat blat /fdb/genome/hg19/chr10.fa indir/gi_615764_gb_T33666 -maxGap=3 outdir/gi_615764_gb_T33666.blat blat /fdb/genome/hg19/chr10.fa indir/gi_615765_gb_T33667 -maxGap=3 outdir/gi_615765_gb_T33667.blatIf you have 1000 input files and therefore 1000 lines, you certainly don't want to create this swarm command file by hand -- the risk of typos would be very large. Instead, write a script to create the swarm command file. This script can be written in bash, perl, python or any language of your choice.
Here is a sample bash script that creates a swarm command file to run Novoalign (a genomics alignment program) for each sequence file in a directory. At the end of the script it submits the swarm.
#!/bin/bash
# this script is called make_swarmfile.sh
# Sample bash script to create a swarm command file and submit it to the Biowulf batch system
# This file creates a swarmfile to run a genomics alignment program on every sequence file in a directory.
# After the swarmfile is created, it submits the swarm
#
# Run this script with: sh make_swarmfile.sh
#
# Swarm docs at https://hpc.nih.gov/apps/swarm.html
# Remember! Before submitting a swarm, you should have checked
# (a) the memory and CPUs required for a single command
# (b) the total disk space (temporary files, output files) required for a single command
# (c) and therefore the total disk space required for the whole swarm. Do you have enough disk space?
# Use 'checkquota' to check!
# you will need to modify the two paths below
inputDir=/path/to/my/input/dir
outputDir=/path/to/my/output/dir
# loop over each file and align against hg19 using Novoalign
for file in ${inputDir}/*
do
filename=`basename $file`
echo "working on file $filename"
echo "novoalign -c \$SLURM_CPUS_PER_TASK -f ${inputDir}/$filename -d /fdb/novoalign/chr_all_hg19.nbx -o SAM > ${outputDir}/$filename.sam" >> novo_swarm.sh
done
At this point, you should check the generated swarm command file (novo_swarm.sh in this case) for sanity. If all looks well, submit with
swarm -f novo_swarm.sh -g # -t #where '-g # ' is the number of GigaBytes of memory required for each swarm command, and '-t #' is the number of threads that you want each instance of the program to run. In this example, Novoalign can multithread successfully to 8 threads, and previous tests have shown that it requires 4 GB of memory, so one would submit with
swarm -f novo_swarm.sh -g 4 -t 8
Please see https://hpc.nih.gov/nih/codex.html (NIH-only).