SAS on Biowulf
 Base SAS provides a scalable, integrated software environment specially designed for data access, transformation and reporting. It includes a fourth-generation programming language; ready-to-use programs for data manipulation, information storage and retrieval, descriptive statistics and report writing; and a powerful macro facility that reduces programming time and maintenance issues. The Base SAS windowing environment provides a full-screen facility for interacting with all parts of a SAS program. On-line help is also available.
Base SAS provides a scalable, integrated software environment specially designed for data access, transformation and reporting. It includes a fourth-generation programming language; ready-to-use programs for data manipulation, information storage and retrieval, descriptive statistics and report writing; and a powerful macro facility that reduces programming time and maintenance issues. The Base SAS windowing environment provides a full-screen facility for interacting with all parts of a SAS program. On-line help is also available.
With multithreaded capabilities, Base SAS software can take advantage of parallel processing power to maximize use of computing resources. However, the main advantage of using SAS on Biowulf would be to run many SAS jobs simultaneously in batch mode as a 'swarm' of single-threaded jobs.
SAS on Biowulf is a limited resource. SLURM keeps track of the SAS resource. Jobs requiring SAS will be started or remain in the queue depending on whether there are SAS resources available. All SAS jobs must therefore specify the resource when the job is submitted, as in the examples below. At present there is a limit of 48 simultaneous SAS processes, but this number is subject to change.
Documentation
- Type 'man sas' at the Biowulf prompt for the SAS man page.
- SAS has online help. In a graphical session, typing 'sas' at the interactive session prompt will bring up the SAS Workspace X windows interface. Click on the Help
button to view the SAS help.
- SAS version 9.4
documentation at sas.com
Important Notes
This application requires a graphical connection. We recommend using a Graphical Session on HPC OnDemand
- Module Name: SAS (see the modules page for more information)
- There is a limit of 48 SAS licenses running simultaneously on the cluster.
- SAS requires bash or ksh to run properly. If you are a t/csh user, you need to start a bash or ksh shell before loading the SAS module and starting SAS, either with or without the GUI.
- To disable automatic HTML reporting of results, go to "Tools" -> "Options" -> "Preferences". Click on the "Results" tab and uncheck the "Create HTML" option.
- The SAS Program Editor clears submitted code by default. To disable this, go to "Tools" -> "Options" -> "Program Editor". Click on the "Editing" tab and uncheck the option "Clear text on submit"
- If you run into an error that suggests "insufficient memory", set the option -memsize when starting SAS. For example sas -memsize 8G.
SAS Products Included
With the latest SAS version included on biowulf, version 9.4M6, we provide all products part of the NIH ISDP enterprise license agreement with SAS. This includes the following Analytical Products:
- SAS/STAT
- SAS/ETS
- SAS/IML
- SAS/QC
SAS Work Directory
By default, SAS on Biowulf is set up to use local disk on the node as the SAS Work directory. You should therefore allocate local disk when submitting a SAS job, or starting an interactive session where you plan to run SAS, as in the examples below. Add --gres=lscratch:# to your sbatch or sinteractive command line to allocate # GB of local disk. For example:
sbatch --gres=lscratch:5 sasjob.sh (allocate 5 GB of local disk for this job)
sinteractive --gres=lscratch:5 (allocate 5 GB of local disk for this interactive session)
More about local disk allocation on Biowulf
Using lscratch for the SAS work directory is by far the most efficient for your jobs.
If you must use another location for the SAS work directory, you will need to add -WORK /path/to/dir to the sas command line. For example:
[user@cn3144 ~]$ sas -nodms -WORK /data/$USER/sastmp/
Do not use /scratch for the work directory. /scratch is a low-performing filesystem and your jobs will run slower.
More about the SAS Work directory
Interactive job
Interactive jobs should be used for debugging, graphics, or applications that cannot be run as batch jobs.
Allocate an interactive session and run the program.
Please do not run SAS on the login node.
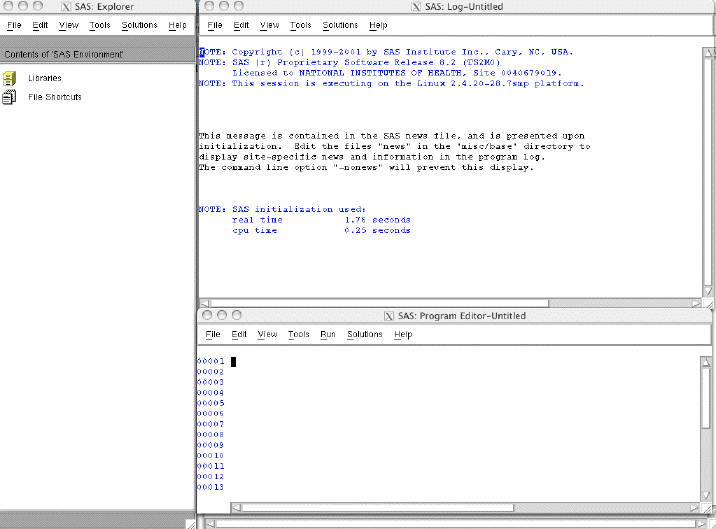
You should see the SAS logo pop up briefly, and then the menus will appear.
Sample session (user input in bold):
[user@biowulf]$ sinteractive --license=sas --gres=lscratch:5
salloc.exe: Pending job allocation 46116226
salloc.exe: job 46116226 queued and waiting for resources
salloc.exe: job 46116226 has been allocated resources
salloc.exe: Granted job allocation 46116226
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn3144 are ready for job
[user@cn3144 ~]$ module load SAS
[+] Loading SAS 9.4 ...
[user@cn3144 ~]$ sas

To use the command-line version of SAS, add -nodms to the command line. e.g.
[user@cn3144 ~]$ sas -nodms
When you're done with your interactive SAS session, exit from the session.
[user@cn3144 ~]$ exit
salloc.exe: Relinquishing job allocation 46116226
[user@biowulf ~]$
Batch job
Create a batch input file (e.g. SAS.sh). For example:
#!/bin/bash
#
#SBATCH -J SAS
#SBATCH --mail-type=BEGIN,END
module load SAS
date
sas sasfile.sas
Submit this job using the Slurm sbatch command.
sbatch --license=sas --gres=lscratch:10 [--cpus-per-task=#] [--mem=#] SAS.sh
This sbatch command requests one SAS license ('license=sas'), so the job will remain in the queue until a license is available. To use more than one license, append --license=sas with a colon and the number of licenses (max is 48) you need (--license=sas:4). You will receive email when the job is executed and when it ends, because of the line containing --mail-type in the script. The output can be found in the directory from which your script was ran, with the file name slurm-#.out
There are other options that you may need to set for your job, specifically walltime and memory. For more information about those options, see
the Biowulf User Guide.
Swarm of Jobs
A
swarm of jobs is an easy way to submit a set of independent commands requiring identical resources.
Create a swarmfile (e.g. SAS.swarm). For example:
sas sasjob1.sas
sas sasjob2.sas
sas sasjob3.sas
sas sasjob4.sas
sas sasjob5.sas
[...]
sas sasjob100.sas
Submit this job using the swarm command.
swarm -f SAS.swarm [-g #] [-t #] --module SAS --gres=lscratch:5 --sbatch "--license=sas"
where
| -g # | Number of Gigabytes of memory required for each process (1 line in the swarm command file)
|
| -t # | Number of threads/CPUs required for each process (1 line in the swarm command file).
|
| --module SAS | Loads the SAS module for each subjob in the swarm
|
| --gres=lscratch:5 | allocates 5 GB of local disk
|
SAS Parallelization
(Thanks to Bruce Swihart, NIAID, for the NTHREADS example below)
As per the SAS documentation, some threading (parallelization using threads on multiple CPUs of a single node) is automatic. The thread-enabled SAS procedures are:
ADAPTIVEREG
FMM
GLM
GLMSELECT
LOESS
MIXED
QUANTLIFE
QUANTREG
QUANTSELECT
ROBUSTREG
Some SAS procedures have a parameter NTHREADS. If set to -1, this will use the number of hyperthreaded cores available on the system. If this parameter is used, the entire node must be allocated exclusively to the job. For example, to use the SAS 'proc nlmixed' procedure, the SAS code could include the following line:
proc nlmixed data=dataset NTHREADS=-1;
The job would then be submitted with
sbatch --exclusive --mem=XG --time=HH:MM:SS --license=sas --gres=lscratch:10 myjob.sh
where
| --exclusive | allocate the entire node (i.e. all CPUs available on the node) for this job
|
| --mem=XG | allocate X gigabytes to the job
|
| --time=HH:MM:SS | set the walltime for this job to HH hours, MM minutes, SS seconds
|
| --license=sas | allocate a SAS license to this job
|
| --gres=lscratch:10 | allocate 10 GB of local disk on the node for this job
|