Anvi’o is an open-source, community-driven analysis and visualization platform for microbial ‘omics. It brings together many aspects of today’s cutting-edge strategies including genomics, metagenomics, metatranscriptomics, pangenomics, metapangenomics, phylogenomics, and microbial population genetics in an integrated and easy-to-use fashion through extensive interactive visualization capabilities.
Allocate an interactive session and run the program.
Sample session (user input in bold):
[user@biowulf ~]$ sinteractive -c4 --mem=8g --gres=lscratch:10 --tunnel
salloc.exe: Pending job allocation 5852147
salloc.exe: job 5852147 queued and waiting for resources
salloc.exe: job 5852147 has been allocated resources
salloc.exe: Granted job allocation 5852147
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn0866 are ready for job
srun: error: x11: no local DISPLAY defined, skipping
Created 1 generic SSH tunnel(s) from this compute node to
biowulf for your use at port numbers defined
in the $PORTn ($PORT1, ...) environment variables.
Please create a SSH tunnel from your workstation to these ports on biowulf.
On Linux/MacOS, open a terminal and run:
ssh -L 42985:localhost:42985 user@biowulf.nih.gov
For Windows instructions, see https://hpc.nih.gov/docs/tunneling
[user@cn0866 ~]$ cd /lscratch/$SLURM_JOB_ID
[user@cn0866 5852147]$ git clone https://github.com/merenlab/anvio.git
Cloning into 'anvio'...
remote: Enumerating objects: 140, done.
remote: Counting objects: 100% (140/140), done.
remote: Compressing objects: 100% (94/94), done.
remote: Total 73821 (delta 79), reused 83 (delta 46), pack-reused 73681
Receiving objects: 100% (73821/73821), 363.99 MiB | 38.97 MiB/s, done.
Resolving deltas: 100% (56214/56214), done.
[user@cn0866 5852147]$ cd anvio/anvio/tests/sandbox/
[user@cn0866 sandbox]$ module load anvio
[+] Loading anvio 7 on cn0866
[+] Loading singularity 3.7.0 on cn0866
[user@cn0866 sandbox]$ anvi-gen-contigs-database -f contigs.fa -o contigs.db \
-n 'An example contigs database'
source: /opt/conda/envs/anvio/etc/conda/activate.d/activate-binutils_linux-64.sh:8:18: parameter expansion requires a literal
source: /opt/conda/envs/anvio/etc/conda/activate.d/activate-gcc_linux-64.sh:8:18: parameter expansion requires a literal
source: /opt/conda/envs/anvio/etc/conda/activate.d/activate-gfortran_linux-64.sh:8:18: parameter expansion requires a literal
source: /opt/conda/envs/anvio/etc/conda/activate.d/activate-gxx_linux-64.sh:8:18: parameter expansion requires a literal
Input FASTA file .............................: /lscratch/5852147/anvio/anvio/tests/sandbox/contigs.fa
Name .........................................: An example contigs database
Description ..................................: No description is given
Num threads for gene calling .................: 1
Finding ORFs in contigs
===============================================
Genes ........................................: /tmp/tmptoj48ad2/contigs.genes
Amino acid sequences .........................: /tmp/tmptoj48ad2/contigs.amino_acid_sequences
Log file .....................................: /tmp/tmptoj48ad2/00_log.txt
CITATION
===============================================
Anvi'o will use 'prodigal' by Hyatt et al (doi:10.1186/1471-2105-11-119) to
identify open reading frames in your data. When you publish your findings,
please do not forget to properly credit their work.
Result .......................................: Prodigal (v2.6.3) has identified 51 genes.
CONTIGS DB CREATE REPORT
===============================================
Split Length .................................: 20,000
K-mer size ...................................: 4
Skip gene calling? ...........................: False
External gene calls provided? ................: False
Ignoring internal stop codons? ...............: False
Splitting pays attention to gene calls? ......: True
Contigs with at least one gene call ..........: 6 of 6 (100.0%)
Contigs database .............................: A new database, contigs.db, has been created.
Number of contigs ............................: 6
Number of splits .............................: 6
Total number of nucleotides ..................: 57,030
Gene calling step skipped ....................: False
Splits broke genes (non-mindful mode) ........: False
Desired split length (what the user wanted) ..: 20,000
Average split length (what anvi'o gave back) .: (Anvi'o did not create any splits)
[user@cn0866 sandbox]$ anvi-run-hmms -c contigs.db
source: /opt/conda/envs/anvio/etc/conda/activate.d/activate-binutils_linux-64.sh:8:18: parameter expansion requires a literal
source: /opt/conda/envs/anvio/etc/conda/activate.d/activate-gcc_linux-64.sh:8:18: parameter expansion requires a literal
source: /opt/conda/envs/anvio/etc/conda/activate.d/activate-gfortran_linux-64.sh:8:18: parameter expansion requires a literal
source: /opt/conda/envs/anvio/etc/conda/activate.d/activate-gxx_linux-64.sh:8:18: parameter expansion requires a literal
HMM sources ..................................: Archaea_76, Bacteria_71, Protista_83,
Ribosomal_RNA_12S, Ribosomal_RNA_16S,
Ribosomal_RNA_18S, Ribosomal_RNA_23S,
Ribosomal_RNA_28S, Ribosomal_RNA_5S
Alphabet/context target found ................: AA:GENE
Alphabet/context target found ................: RNA:CONTIG
HMM Profiling for Archaea_76
===============================================
Reference ....................................: Lee, https://doi.org/10.1093/bioinformatics/btz188
[snip...]
HMM Profiling for Ribosomal_RNA_5S
===============================================
Reference ....................................: Seeman T, https://github.com/tseemann/barrnap
Kind .........................................: Ribosomal_RNA_5S
Alphabet .....................................: RNA
Context ......................................: CONTIG
Domain .......................................: N/A
HMM model path ...............................: /tmp/tmppgigaeov/Ribosomal_RNA_5S.hmm
Number of genes in HMM model .................: 5
Noise cutoff term(s) .........................: --cut_ga
Number of CPUs will be used for search .......: 1
HMMer program used for search ................: nhmmscan
Temporary work dir ...........................: /tmp/tmpzt2y6k5k
Log file for thread 0 ........................: /tmp/tmpzt2y6k5k/RNA_contig_sequences.fa.0_log
Number of raw hits ...........................: 0
* The HMM source 'Ribosomal_RNA_5S' returned 0 hits. SAD (but it's stil OK).
[user@cn0866 sandbox]$ anvi-display-contigs-stats contigs.db -P $PORT1
source: /opt/conda/envs/anvio/etc/conda/activate.d/activate-binutils_linux-64.sh:8:18: parameter expansion requires a literal
source: /opt/conda/envs/anvio/etc/conda/activate.d/activate-gcc_linux-64.sh:8:18: parameter expansion requires a literal
source: /opt/conda/envs/anvio/etc/conda/activate.d/activate-gfortran_linux-64.sh:8:18: parameter expansion requires a literal
source: /opt/conda/envs/anvio/etc/conda/activate.d/activate-gxx_linux-64.sh:8:18: parameter expansion requires a literal
* The server is up and running \U0001f389
WARNING
===============================================
If you are using OSX and if the server terminates prematurely before you can see
anything in your browser, try running the same command by putting 'sudo ' at the
beginning of it (you will be prompted to enter your password if sudo requires
super user credentials on your system). If your browser does not show up, try
manually entering the URL shown below into the address bar of your favorite
browser. *cough* CHROME *cough*.
Server address ...............................: http://0.0.0.0:42985
* When you are ready, press CTRL+C once to terminate the server and go back to the
command line.
To view the server, you will need to establish an ssh tunnel between your local workstation and the biowulf login node. In a new window:
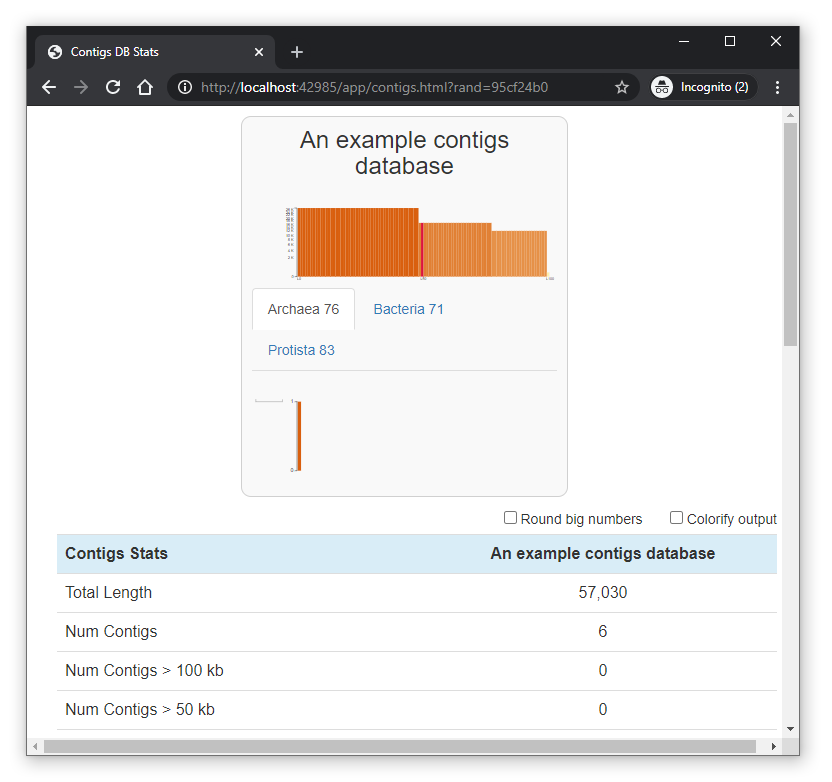
[user@my_workstation ~]$ ssh -L 42985:localhost:42985 <username>@biowulf.nih.govYou can now point your local browser to localhost:<port> where <port> refers to the port you were assigned when you initiated your session. You should see a window like that pictured below:
Create a batch input file (e.g. anvio.sh). For example:
#!/bin/bash
set -e
module load anvio
cd /data/${USER}/path/to/data
anvi-init-bam SAMPLE-01-RAW.bam -o SAMPLE-01.bam
Submit this job using the Slurm sbatch command.
sbatch [--cpus-per-task=#] [--mem=#] anvio.sh
Create a swarmfile (e.g. anvio.swarm). For example:
anvi-init-bam SAMPLE-01-RAW.bam -o SAMPLE-01.bam anvi-init-bam SAMPLE-02-RAW.bam -o SAMPLE-02.bam anvi-init-bam SAMPLE-03-RAW.bam -o SAMPLE-03.bam anvi-init-bam SAMPLE-04-RAW.bam -o SAMPLE-04.bam
Submit this job using the swarm command.
swarm -f anvio.swarm [-g #] [-t #] --module anviowhere
| -g # | Number of Gigabytes of memory required for each process (1 line in the swarm command file) |
| -t # | Number of threads/CPUs required for each process (1 line in the swarm command file). |
| --module anvio | Loads the anvio module for each subjob in the swarm |