This extensive package of tools is being developed by Brian Bushnell. It includes
and many more.
On biowulf all bbtools are used through a wrapper script that automatically passes correct memory information to each of the tools. This is important as the automatic memory detection by the individual tools is not slurm aware and reports incorrect amounts of available memory.
Usage of the wrapper script:
NAME
bbtools - BBMap short read aligner and other bioinformatic tools
SYNOPSIS
bbtools command [options]
DESCRIPTION
bbtools is a convenient frontend to the collection of bioinformatic
tools created by Brian Bushnell at the Joint Genome Institute. It
includes a short read mapper, a k-mer based normalization tool, refor-
matting tools, and many more. The wrapper script will automatically set
an appropriate maximum memory for the JVM executing the code. It will
limit runs on helix to 10GB and use SLURM to determine the correct
amount of memory for batch/interactive jobs.
COMMANDS
help display this help message
man list all available commands with their descrcription and gen-
eral usage information
list list all available commands
All other commands are essentially the name of the corresponding
bbtools script without the extension.
OPTIONS
Options are tool specific. See tool documentation for more details.
use --help to get tool specific help.
AUTHOR OF WRAPPER SCRIPT
Wolfgang Resch. Contact staff@hpc.nih.gov for help.
Use bbtools man to see an overview of all available tools as well
as some general documentation.
Allocate an interactive session and run the program. Sample session:
[user@biowulf]$ sinteractive
salloc.exe: Pending job allocation 46116226
salloc.exe: job 46116226 queued and waiting for resources
salloc.exe: job 46116226 has been allocated resources
salloc.exe: Granted job allocation 46116226
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn3144 are ready for job
[user@cn3144 ~]$ module load bbtools
[user@cn3144 ~]$ r1=/usr/local/apps/bbtools/TEST_DATA/read1_2000k.fastq.gz
[user@cn3144 ~]$ r2=/usr/local/apps/bbtools/TEST_DATA/read2_2000k.fastq.gz
[user@cn3144 ~]$ bbtools reformat in=${r1} in2=${r2} \
out=read12_2000k.fastq.gz \
qin=auto qout=33 ow=t cardinality=t \
bhist=read12.bhist qhist=read12.qhist lhist=read12.lhist
[user@cn3144 ~]$ exit
salloc.exe: Relinquishing job allocation 46116226
[user@biowulf ~]$
If you would like to specify the Java memory usage, you may add a custom -Xmx# to your command. The wrapper will set
the maximum JVM heap size to 85% of what you select. (See the -Xmx17408m below).
[user@biowulf ~]# bbtools bbmap -Xmx20g java -ea -Xmx17408m -Xms17408m -cp /usr/local/apps/bbtools/38.87/bbmap_src/current/ align2.BBMap build=1 overwrite=true fastareadlen=500 -Xmx17408m Executing align2.BBMap [build=1, overwrite=true, fastareadlen=500, -Xmx17408m] Version 38.87 No output file. [...]
As an example for using a few of the tools we will download some bacterial genomes, simulate reads from these genomes in different ratios, apply kmer based normalization, align the raw and normalized reads to the genomes, and finally plot the coverage of the genomes before and after normalization.
This is all included in a single script for this example. For a real analysis, each of these steps should be carried out separately.
#! /bin/bash
ENSBACT="ftp://ftp.ensemblgenomes.org/pub/bacteria/release-30/fasta/bacteria"
module load bbtools || exit 1
module load R || exit 1
################################################################################
# copy bacterial genomes #
################################################################################
# from the TEST_DATA directory
mkdir -p data tmp out
cp /usr/local/apps/bbtools/TEST_DATA/drad.fa.gz data # Deinococcus radiodurans
dr=data/drad.fa.gz
cp /usr/local/apps/bbtools/TEST_DATA/ecoli.fa.gz data # Eschericia coli
ec=data/ecoli.fa.gz
cp /usr/local/apps/bbtools/TEST_DATA/saur.fa.gz data # Staphylococcus aureus
sa=data/saur.fa.gz
################################################################################
# simulate reads #
################################################################################
# simulate 50 nt reads with different coverage from each of the genomes. the
# amp parameter simulates amplification.
bbtools randomreads ref=$ec build=1 out=tmp/ec.fq length=50 \
reads=2000000 seed=132565 snprate=0.05 amp=50
bbtools randomreads ref=$dr build=2 out=tmp/dr.fq length=50 \
reads=4000000 seed=3565 snprate=0.05 amp=200
bbtools randomreads ref=$sa build=3 out=tmp/sa.fq length=50 \
reads=12000000 seed=981 snprate=0.05 amp=200
cat tmp/ec.fq tmp/dr.fq tmp/sa.fq | gzip -c > data/mg_raw.fq.gz
rm tmp/ec.fq tmp/dr.fq tmp/sa.fq
################################################################################
# kmer normalization #
################################################################################
# normalize coverage and do error correction
bbtools bbnorm in=data/mg_raw.fq.gz out=data/mg_norm.fq.gz tmpdir=./tmp threads=$SLURM_CPUS_PER_TASK \
target=20 k=25 ecc=t mindepth=2
################################################################################
# align raw and normalized against the three genomes #
################################################################################
bbtools bbsplit build=4 ref_ec=$ec ref_dr=$dr ref_sa=$sa
bbtools bbsplit build=4 in=data/mg_raw.fq.gz out_ec=out/mg_raw_ec.bam \
out_dr=out/mg_raw_dr.bam out_sa=out/mg_raw_sa.bam refstats=out/mg_raw_stat \
t=$SLURM_CPUS_PER_TASK
bbtools bbsplit build=4 in=data/mg_norm.fq.gz out_ec=out/mg_norm_ec.bam \
out_dr=out/mg_norm_dr.bam out_sa=out/mg_norm_sa.bam refstats=out/mg_norm_stat \
t=$SLURM_CPUS_PER_TASK
################################################################################
# calculate coverage for all 6 samples #
################################################################################
for f in out/mg_*.bam; do
bbtools pileup in=$f out=${f%.bam}.covstat hist=${f%.bam}.covhist
done
# merge into a single file
rm -f out/mg_covhist.csv
for f in out/mg_*.covhist; do
fld=( $(echo $f | tr '_.' ' ') )
awk -v t=${fld[1]} -v o=${fld[2]} 'NR > 1 {{print $1","$2","t","o}}' $f \
>> out/mg_covhist.csv
done
# plot the coverage data
echo > tmp/plot.R <<EOF
library(ggplot2)
dta <- read.table("out/mg_covhist.csv", sep=",", col.names=c("cov", "freq", "type", "org"))
dta <- subset(dta, cov < 400)
p <- ggplot(dta) +
geom_line(aes(x=cov, y=freq, col=org), size=1) +
scale_color_brewer(palette="Set1") +
facet_wrap(~type, nrow=2, scale="free_x") +
theme_bw(14) +
theme(legend.position="top", panel.grid.minor=element_blank(),
panel.grid.major=element_blank(), panel.border=element_rect(colour="black"))
ggsave(p, file="out/mg_covhist.png")
EOF
Rscript tmp/plot.R
Submit this job using the Slurm sbatch command.
sbatch --mem=10G --cpus-per-task=6 examble.sh
This generates the following graphs demonstrating the effect of kmer based normalization:
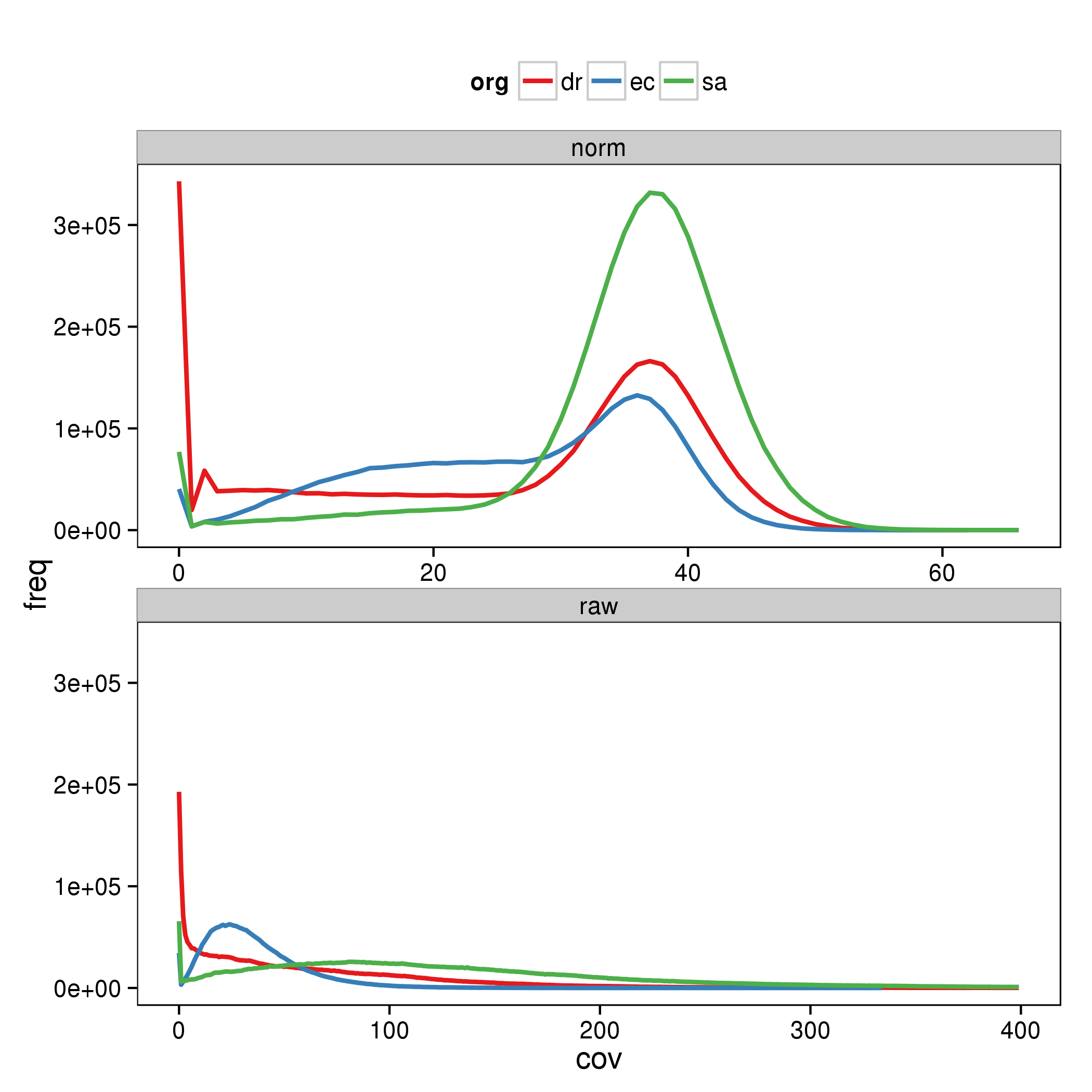
Create a swarmfile (e.g. bbtools.swarm). For example:
bbtools reformat in=a1.fq.gz in2=a2.fq.gz \ out=a12.fq.gz \ qin=auto qout=33 ow=t cardinality=t bbtools reformat in=b1.fq.gz in2=b2.fq.gz \ out=b12.fq.gz \ qin=auto qout=33 ow=t cardinality=t bbtools reformat in=c1.fq.gz in2=c2.fq.gz \ out=c12.fq.gz \ qin=auto qout=33 ow=t cardinality=t
Submit this job using the swarm command.
swarm -f bbtools.swarm -g 4 -t 2 --module bbtoolswhere
| -g # | Number of Gigabytes of memory required for each process (1 line in the swarm command file) |
| -t # | Number of threads/CPUs required for each process (1 line in the swarm command file). |
| --module bbtools | Loads the bbtools module for each subjob in the swarm |