CHARMM (Chemistry at HARvard Macromolecular Mechanics) [1]:
In order provide support for multiple executable types using a range of parallel communication methods and hardware via a common interface, cover scripts have been made available in /usr/local/apps/charmm/bin on Biowulf, each named for the CHARMM release version they support. The recommended setup is to use
module load charmm
to modify the command search path for your shell (bash or csh); the cover script will load any additional modules required. Besides the cover scripts, a tool for extracting data from CHARMM output log files, getprop.csh, is also available. Descriptions of the cover script commands, syntax listings, and usage examples are given in this section. General usage notes for running CHARMM via the SLURM queue and using the X11 graphics are given below, after the cover script descriptions.
biowulf /<6>charmm [5] module load charmm
biowulf /<6>charmm [6] c42b2 -help
Syntax; square brackets indicate [ optional args ]
SINGLE PROC
c42b2 [options] [ charmm-args ] < file.inp >& file.out
Omit "< file.inp >& file.out" for interactive use (e.g. graphics)
PARALLEL
c42b2 [options] ompi Nproc file.inp [ charmm-args ] >& file.out # OpenMPI
c42b2 -h | -help # this listing
Notes:
[options] ; must precede parallel keywords, order dependent
verbose :: prints additional environment info; must be *first*
ddg :: domdec_gpu; requires node with GPU (-p gpu)
sse :: override AVX architecture detection
PM Ewald type override option; default includes COLFFT and DOMDEC:
async :: alt. (slower) PME method, incl. REPDSTR and MSCALE
Parallel args; input filename required as 3rd arg
ompi :: use the OpenMPI parallel library
Nproc :: number of MPI processes; one per core, or one per GPU (ddg)
charmm-args :: optional script @ vars, in the form N:27 or RUN=15 etc.
(N.B. must follow any options and parallel args)
Examples:
c42b2 MDL:2 < minmodel.inp >& minmodel.out # single proc min
c42b2 ompi 16 minmodel.inp MDL:2 >& minmodel.out # minimization
c42b2 ompi 64 dyn.inp >& dyn.out # COLFFT or DOMDEC
c42b2 ddg ompi 8 dyn.inp >& dyn.out # DOMDEC_GPU
c42b2 ompi 64 dyn.inp -chsize 450000 >& dyn.out # 450000 atom limit
c42b2 sse ompi 64 dyn.inp >& dyn.out # force use of SSE on AVX host
c42b2 async ompi 32 dyn.inp >& dyn.out # async; P21 symmetry, REPDSTR
The above usage examples illustrate the positional keywords; parallel usage requires the ompi keyword, followed by two more arguments for the number of cores (not SLURM cpus!) and finally the input file name. The optional mutually exclusive arguments ddg and async invoke different executables, compiled with different feature sets and with different run-time library requirements; the c42b2 cover script loads the modules needed, e.g. CUDA for ddg, and then invokes the requested executable. The async keyword includes support for features such as replica exchange MD, non-orthogonal crystal lattices, simulation of the assymetric unit with rotations of the simulation cell, and a number of other custom features; the fast DOMDEC code is NOT supported.
The sse keyword uses an older version of the Intel chipset floating point microarchitecture instructions, and is mainly used for testing.
Which options to use depends on the details of the types of calculations or operations being done, the type of molecular system, the boundary conditions, and probably other factors.
For a non-parallel CHARMM job such as model building or ad hoc trajectory analysis, the commands and setup have few requirements. The job script (build.csh) can be simply:
#!/bin/csh cd $SLURM_SUBMIT_DIR module load charmm c42b2 < build-psf.inp >& build-psf.out
The above can be submitted to the batch queue via:
sbatch build.csh
For parallel usage, the following script (sbatch.csh) illustrates submitting a SLURM batch job which will use the 16 physical cores on each of 4 nodes (64 total cores:
#!/bin/csh # use subdir name for job id and log file names set id = $cwd:t # nodes @ n = 4 # tasks, for 16 core nodes @ nt = 16 * $n sbatch --ntasks=$nt -J $id -o $id.%j job.csh
Assuming the Infiniband (multinode) nodes will be used, the job.csh script contains:
#!/bin/csh #SBATCH --partition=multinode #SBATCH --exclusive #SBATCH --ntasks-per-core=1 cd $SLURM_SUBMIT_DIR module load charmm c42b2 ompi $SLURM_NTASKS charmmrun.inp >& charmmrun.out
The environment variable SLURM_SUBMIT_DIR points to the working directory where 'sbatch' was run, and SLURM_NTASKS contains the value given with the --ntasks= argument to sbatch. The above is suitable for most parallel CHARMM usage, other than the DOMDEC_GPU code invoked via the ddg cover script keyword (see below). A more detailed example for long simulations, also using separate scripts for job submission and job execution, is given in the Daisy Chaining section below. Of course, one could submit job.csh directly via e.g.:
sbatch --ntasks=64 -J MyJob -o MyJob.%j job.csh
where %j represents the SLURM job number.
The DOMDEC_GPU code invoked via the ddg cover script keyword uses both an MPI library (OpenMPI in this case) and OpenMP threads, and therefore requires changes to the SLURM sbatch arguments. The changes are shown in the example below (sbatchGPU.csh); the --nodes= argument must be included, and the --ntasks= value must be twice the number of nodes, as each node has two GPU devices.
#!/bin/csh # use subdir name for job id and log file names set id = $cwd:t # nodes @ n = 4 # tasks, for nodes with 2 GPUs @ nt = 2 * $n # use all eight GPUs on four nodes sbatch --ntasks=$nt --nodes=$n -J $id -o $id.%j gpujob.csh
A minimal job script follows, which illustrates additional changes to the information given to SLURM, via directives that do not depend on the number of nodes requested. Other than requesting --exclusive use of the node, all of the directives are distinct from those in the job.csh example above for general parallel usage.
#!/bin/csh #SBATCH --partition=gpu #SBATCH --exclusive #SBATCH --cpus-per-task=8 #SBATCH --gres=gpu:k20x:2 cd $SLURM_SUBMIT_DIR module load charmm c42b2 ddg ompi $SLURM_NTASKS charmmgpu.inp >& charmmgpu.out
For a variety of tasks such as model building, analysis, and graphics, foreground interactive use of CHARMM can be advantageous, esp. when developing and testing a new input script. The SLURM sinteractive command makes this fairly easy (system prompts in bold, user input in italics):
biowulf /<2>EwaldNVE [69] sinteractive salloc.exe: Granted job allocation 1693180 salloc.exe: Waiting for resource configuration salloc.exe: Nodes cn0032 are ready for job cn0032 /<2>EwaldNVE [1] module load charmm cn0032 /<2>EwaldNVE [2] c42b2 < build-psf.inp >& build-psf.out cn0032 /<2>EwaldNVE [3] exit exit salloc.exe: Relinquishing job allocation 1693180 salloc.exe: Job allocation 1693180 has been revoked.
By adding a couple SLURM options, one can also run parallel minimization jobs as well:
biowulf /<2>EwaldNVE [70] sinteractive -n 8 --ntasks-per-core=1 salloc.exe: Granted job allocation 1693189 salloc.exe: Waiting for resource configuration salloc.exe: Nodes cn0254 are ready for job cn0254 /<2>EwaldNVE [1] module load charmm cn0254 /<2>EwaldNVE [2] c42b2 < build-psf.inp >& build-psf.out cn0254 /<2>EwaldNVE [3] c42b2 ompi 8 minmodel.inp >& minmodel.out & cn0254 /<2>EwaldNVE [4] exit exit salloc.exe: Relinquishing job allocation 1693189
For troubleshooting, it may be useful to pipe the output, and both save it in file (via 'tee') and view it in the 'less' browser, e.g.:
cn0254 /<2>EwaldNVE [3] c42b2 ompi 8 minmodel.inp |& tee minmodel.out | less
Finally, CHARMM itself can be run interactively, via simply:
biowulf /<2>EwaldNVE [71] sinteractive
salloc.exe: Granted job allocation 1693592
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn0103 are ready for job
cn0103 /<2>EwaldNVE [1] module load charmm
cn0103 /<2>EwaldNVE [2] c42b2
Linux cn0074 3.10.0-693.2.2.el7.x86_64 #1 SMP Tue Sep 12 22:26:13 UTC 2017 x86_64 x86_64 x86_64 GNU/Linux
16:39:01 up 2 days, 17:06, 0 users, load average: 18.98, 18.69, 18.38
[+] Loading Intel 2018.1.163 Compilers ...
>>>========>> FOR SYNTAX AND NOTES, TRY "c42b2 -help"
-rwxrwxr-x 1 venabler 38816824 Jun 3 16:25 /usr/local/apps/charmm/c42b2/em64t/ifortavx.x11
/usr/local/apps/charmm/c42b2/em64t/ifortavx.x11
1
Chemistry at HARvard Macromolecular Mechanics
(CHARMM) - Developmental Version 42b2 February 15, 2018
Copyright(c) 1984-2014 President and Fellows of Harvard College
All Rights Reserved
Current operating system: Linux-3.10.0-693.2.2.el7.x86_64(x86_64)@cn0074
Created on 6/7/18 at 16:39:01 by user: venabler
Maximum number of ATOMS: 360720, and RESidues: 120240
At this point the program is expecting input, starting with a title; it is recommended to type bomlev -1 as the first command, as that will forgive typing errors and allow the program to continue. It also recommended to have the initial setup commands (reading RTF and PARAM files, PSF and COOR files, etc.) in a 'stream' file, so that those actions can be done vie e.g.
stream init.str
The same applies to other complex setups, such as establishing restraints, or graphics setup.
Note that the graphics uses X11, so the initial login to biowulf should use either the -X or the -Y option of the ssh command, to enable X11 tunneling for the graphics display.
Recent versions of the distributed CHARMM parameters, including the latest release, are available in /usr/local/apps/charmm as subdirectories topparYYYY where YYYY is the release year. Each release contains a number of corrections and additions from the past year, esp. for the CHARMM force fields. The toppar2017 release uses a newer format, so one should be careful about using it with older PSF files. Files distributed with models built using CHARMM-GUI now use this newer format.
In order to run a long series of CHARMM calculations, a systematic method has been developed over many years and operating systems. There are several basic concepts:
#!/bin/csh
#SBATCH --partition=multinode
#SBATCH --exclusive
#SBATCH --ntasks-per-core=1
# ASSUMPTION (1): output files are named dyn.res dyn.trj dyn.out
# ASSUMPTION (2): previous restart file read as dyn.rea
#
cd $SLURM_SUBMIT_DIR
if ( ! -d Res ) mkdir Res
if ( ! -d Out ) mkdir Out
if ( ! -d Crd ) mkdir Crd
if ( ! -d Trj ) mkdir Trj
set chm = "/usr/local/apps/charmm/bin/c42b2 ompi $SLURM_NTASKS"
set nrun = 1
set d = $cwd:t
@ krun = 1
while ( $krun <= $nrun )
if ( -e next.seqno ) then
$chm dyn.inp D:$d > dyn.out
else
$chm dynstrt.inp D:$d > dyn.out
endif
set okay = true
# TEST FOR EXISTENCE, THEN NONZERO LENGTH OF OUTPUT FILES
if ( -e dyn.res && -e dyn.dcd ) then
@ res = `wc dyn.res | awk '{print $1}'`
@ tsz = `ls -s dyn.dcd | awk '{print $1}'`
@ nrm = `grep ' NORMAL TERMINATION ' dyn.out | wc -l`
if ( $res > 100 && $tsz > 0 && $nrm == 1 ) then
# SUCCESSFUL RUN; COPY RESTART FILE
cp dyn.res dyn.rea
# DETERMINE RUN NUMBER
if ( -e next.seqno ) then
@ i = `cat next.seqno`
else
@ i = 1
endif
# NUMBER AND MOVE THE OUTPUT FILES
mv dyn.out Out/dyn$i.out
mv dyn.crd Crd/dyn$i.crd
mv dyn.res Res/dyn$i.res
mv dyn.dcd Trj/dyn$i.dcd
gzip -f Out/dyn$i.out Res/dyn$i.res Crd/dyn$i.crd
# CONDITIONAL END CHECK
if ( -e last.seqno ) then
@ l = `cat last.seqno`
if ( $i == $l ) then
@ i += 1
echo $i > next.seqno
exit
endif
endif
@ i += 1
echo $i > next.seqno
else
# ZERO LENGTH FILE(S)
set okay = false
endif
else
# FILE DOESN'T EXIST
set okay = false
endif
# TEST FOR CHARMM RUN FAILED; CREATE .ERR FILE WITH TIMESTAMP
if ( $okay == true ) then
# SUBMIT THE NEXT JOB
if ( $krun == $nrun ) ./sbatch.csh
@ krun += 1
else
set ts = `date +%m%d.%H%M`
date > msg.tmp
echo $cwd >> msg.tmp
head -64 dyn.out >> msg.tmp
tail -64 dyn.out >> msg.tmp
mv dyn.out dyn.err.$ts
mail -s "$SLURM_JOB_NAME $SLURM_JOB_ID $ts" $USER@helix.nih.gov < msg.tmp
exit(201)
endif
end
The above can be used for most parallel CHARMM usage (except for GPUs); for some types of calculations, the async keyword may be needed as well.
Plain text versions of the scripts are given in the following links; after downloading, rename them to .csh files, and allow them to be executed via e.g.
mv sbatch.txt sbatch.csh chmod u+x sbatch.csh
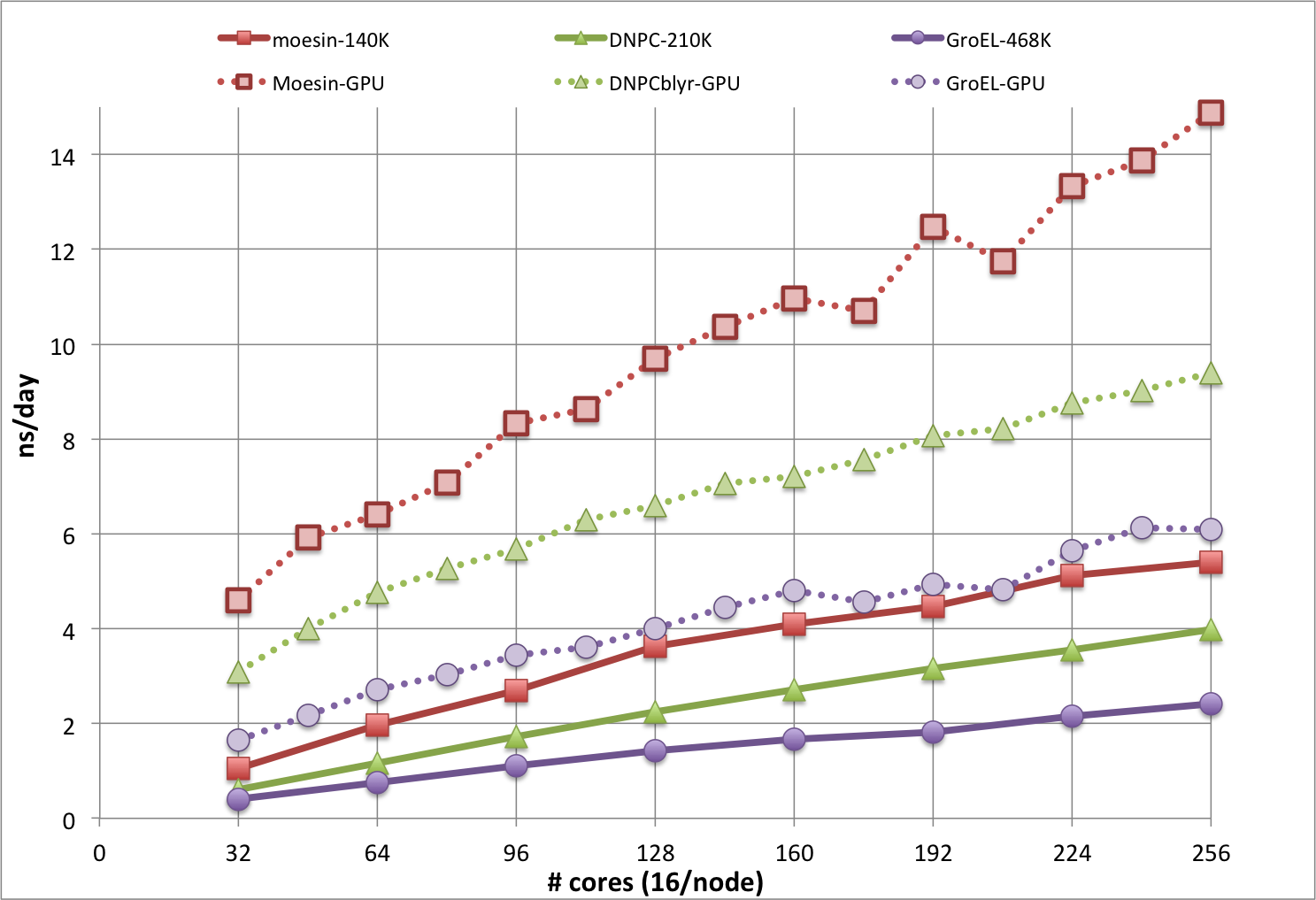
The plot below is from CHARMM benchmarks run during the beta test phase of Biowulf, and shows results for DOMDEC on Infiniband nodes (solid lines) and DOMDEC_GPU on gpu nodes (dotted lines), for even numbers from 2 through 16 nodes. The timings in ns/day are from short MD simulations, run with a 1 fs integration time step, for 3 molecular systems of different sizes and shapes:
Note that the ns/day rate would be doubled with the use of a 2 fs time step, which is often done for more exploratory sampling, but not necessarily recommended for the best accuracy and precision. Simulations systems that cannot use DOMDEC will be somewhat slower, and will not scale well past about 64 cores.