deFuse uses clusters of discordant paired end reads to guide split read alignments across gene-gene fusion boundaries in RNA-Seq data. Filters are applied to reduce false positives and results are annotated.
Reference data sets required by deFuse are stored under
/fdb/defuse Note that the versions of gmap/gsnap available on biowulf make use of a
new index format. Only reference data sets ending in _newgmap are
compatible with these versions of gmap.
deFuse runs are set up using a configuration file which may change between defuse versions. Use the configuration file included with the version you are using as a starting point for your analysis. They can be found under
/usr/local/apps/defuse/[version]/config_*.txt
deFuse is a pipeline that makes use of internal and external tools. Pipeline
steps can be run on the same machine as the main driver script
(defuse.pl -s direct ...) or submitted to compute nodes (
defuse.pl -s slurm ...). The -p option determines how many jobs
are run in parallel.
We do not recommend to use "-s slurm" on biowulf because it will submit multiple short subjobs, which will take longer queue time.
defuse.pl option summary (for version 0.8.0):
Usage: defuse.pl [options]
Run the deFuse pipeline for fusion discovery.
-h, --help Displays this information
-c, --config Configuration Filename
-d, --dataset Dataset Directory
-o, --output Output Directory
-r, --res Main results filename (default: results.tsv
in Output Directory)
-a, --rescla Results with a probability column filename
(default: results.classify.tsv in Output Directory)
-b, --resfil Filtered by the probability threshold results filename
(default: results.filtered.tsv in Output Directory)
-1, --1fastq Fastq filename 1
-2, --2fastq Fastq filename 2
-n, --name Library Name (default: Output Directory Suffix)
-l, --local Job Local Directory (default: Output Directory)
-s, --submit Submitter Type (default: direct)
-p, --parallel Maximum Number of Parallel Jobs (default: 1)
Note that the driver script was renamed to defuse_run.pl
in version 0.8.0. However, defuse.pl is still available
as a symbolic link. Note also that starting with version 0.8.0 the
dataset directory has to be provided on the command line with -d
$DEFUSE_TEST_DATAAllocate an interactive session and run the program. Sample session:
[user@biowulf]$ sinteractive --cpus-per-task=16 --mem=20g salloc.exe: Pending job allocation 46116226 salloc.exe: job 46116226 queued and waiting for resources salloc.exe: job 46116226 has been allocated resources salloc.exe: Granted job allocation 46116226 salloc.exe: Waiting for resource configuration salloc.exe: Nodes cn3144 are ready for job [user@cn3144 ~]$ DATA=/usr/local/apps/defuse/TEST_DATA/small [user@cn3144 ~]$ module load defuse [user@cn3144 ~]$ cp /usr/local/apps/defuse/config_hg19_ens69.txt config.txt [user@cn3144 ~]$ defuse.pl -c config.txt -o small.out \ -d /fdb/defuse/hg19_ens69_newgmap \ -1 $DATA/rna/spiked.1.fastq -2 $DATA/rna/spiked.2.fastq \ -s direct -p 12 [user@cn3144 ~]$ [user@cn3144 ~]$ exit salloc.exe: Relinquishing job allocation 46116226 [user@biowulf ~]$
A deFuse batch job can run in two different ways - either all jobs started
by the main defuse.pl are run on the same compute node, or they
are submitted to other nodes via slurm.
Here is an example script that will run all jobs on the same node as the node running the main deFuse script. This makes use of a small data set of simulated RNASeq reads. Note that bowtie is allowed 2 threads in the sample config file, so the number of parallel jobs is limited to half the number of CPUs.
#! /bin/bash
# filename: small.sh
set -e
DATA=/usr/local/apps/defuse/TEST_DATA/small
DEFUSE_VER=0.8.1
module load defuse/$DEFUSE_VER || exit 1
cp -r $DATA . || exit 1
cp /usr/local/apps/defuse/$DEFUSE_VER/config_hg19_ens69.txt config.txt
defuse.pl -c config.txt -o small.out \
-d /fdb/defuse/hg19_ens69_newgmap \
-1 small/rna/spiked.1.fastq -2 small/rna/spiked.2.fastq \
-s direct -p $SLURM_CPUS_PER_TASK
The batch file is submitted to the queue with a command similar to the following:
biowulf$ sbatch --cpus-per-task=20 small.sh
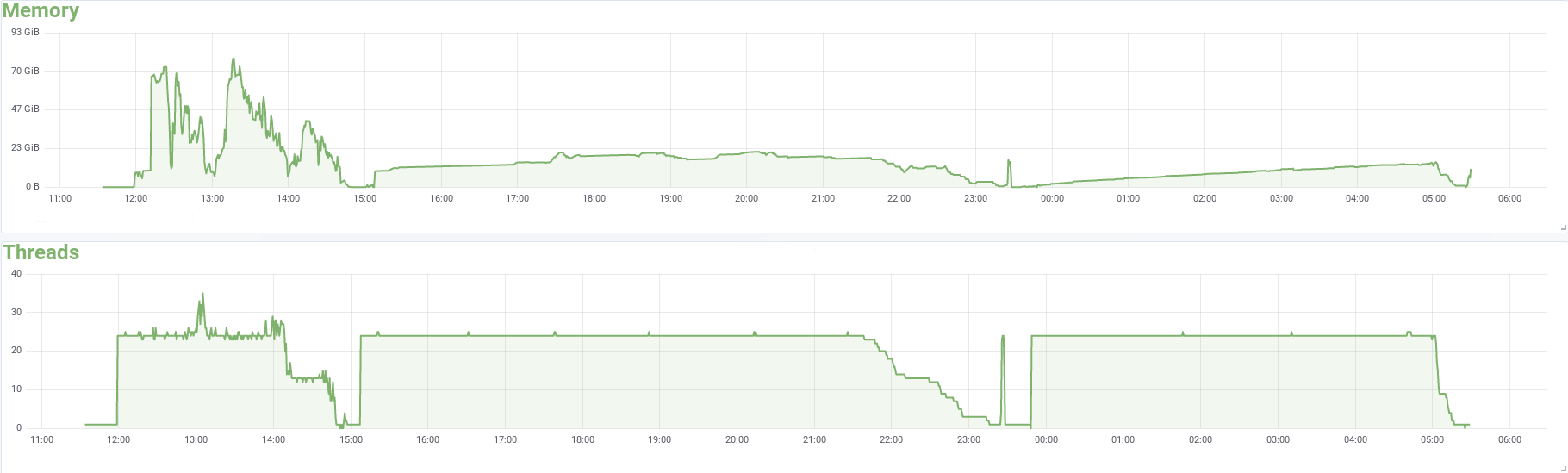
The profile of a local defuse job with 24 CPUs processing 60M 125nt paired end reads:
The other approach is shown in the following batch script with runs the main script on a compute node with just 2 CPUs allocated. The main script in turn submits subjobs via slurm. This example uses data obtained from the Gerstein lab for cell line NCI-H660, which contains a known TMPRSS2-ERG fusion.
#! /bin/bash
# this file is large.sh
# defuse version
DEFUSE_VER=0.8.1
module load defuse/$DEFUSE_VER || exit 1
cp /usr/local/apps/defuse/$DEFUSE_VER/config_hg19_ens69.txt config.txt
# large test data - copy if it doesn't already exist
if [[ ! -d large ]]; then
cd large
DATA=/usr/local/apps/defuse/TEST_DATA/NCIH660
cp $DATA/NCIH660.fastq.tar.gz .
tar -xzf NCIH660.fastq.tar.gz
rm NCIH660.fastq.tar.gz
cd ..
fi
defuse.pl -c config.txt -o ncih660.out \
-d /fdb/defuse/hg19_ens69_newgmap \
-1 large/NCIH660_1.fastq \
-2 large/NCIH660_2.fastq \
-s slurm -p 25
Which is submitted as follows
biowulf$ sbatch large.sh
To set up a swarm of defuse jobs, each running the subjobs in local mode, use a swarm file like this:
defuse.pl -c config.txt -o defuse1.out \ -d /fdb/defuse/hg19_ens69_newgmap \ -1 defuse1.1.fastq \ -2 defuse1.2.fastq \ -s direct -p 12 defuse.pl -c config.txt -o defuse2.out \ -d /fdb/defuse/hg19_ens69_newgmap \ -1 defuse2.1.fastq \ -2 defuse2.2.fastq \ -s direct -p 12 [...]
Then submit the swarm, requesting 24 CPUs and 10GB memory for each task
biowulf$ swarm -g 10 -t 24 swarmfile --module=defuse/0.8.1where
| -g # | Number of Gigabytes of memory required for each process (1 line in the swarm command file) |
| -t # | Number of threads/CPUs required for each process (1 line in the swarm command file). |
| --module defuse | Loads the defuse module for each subjob in the swarm |