
esm-fold run with default settings on 300 mimivirus
proteins. Batch sizes (n) are indicated at the top.Transformer protein language models from the Meta Fundamental AI AI Research Protein Team.
$TORCH_HOME is set to allow esm to find the pretrained models in /fdb on biowulf$ESM_TEST_DATAesm-python python interpreter
and a helper to install a jupyter kernel for the python including the esm module and its
dependencies.$HF_HUB_CACHE to point to your huggingface
cache directory containing the model weights.Allocate an interactive session and run the program. Sample session:
esm-fold is designed to model many proteins in a single run. For the following example
we will model 300 monomers in a single fasta file. Multimers can be modeled by separating chains with a ':'
[user@biowulf]$ sinteractive --gres=lscratch:100,gpu:a100:1 --mem=48g --cpus-per-task=8
salloc.exe: Pending job allocation 46116226
salloc.exe: job 46116226 queued and waiting for resources
salloc.exe: job 46116226 has been allocated resources
salloc.exe: Granted job allocation 46116226
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn3144 are ready for job
[user@cn3144]$ module load esm/2.0.0
[+] Loading esm 2.0.0
[user@cn3144]$ #fetch 300 sequences. esm-fold will process them sorted by length
[user@cn3144]$ awk '/>/{n++}n>300{exit}{print}' $ESM_TEST_DATA/mimi_all.fa > test.fa
[user@cn3144]$ export TMPDIR=/lscratch/$SLURM_JOB_ID
[user@cn3144]$ esm-fold -i test.fa -o test_models
23/04/13 12:48:05 | INFO | root | Reading sequences from test.fa
23/04/13 12:48:05 | INFO | root | Loaded 300 sequences from test.fa
23/04/13 12:48:05 | INFO | root | Loading model
...
23/04/13 12:48:38 | INFO | root | Starting Predictions
23/04/13 12:48:42 | INFO | root | Predicted structure for YP_004021047.1 with length 31, pLDDT 81.5, pTM 0.502 in 0.2s (amortized, batch size 18). 1 / 300 completed.
23/04/13 12:48:42 | INFO | root | Predicted structure for YP_004021044.1 with length 36, pLDDT 62.8, pTM 0.350 in 0.2s (amortized, batch size 18). 2 / 300 completed.
...
23/04/13 12:49:17 | INFO | root | Predicted structure for YP_003987280.1 with length 149, pLDDT 35.6, pTM 0.308 in 1.1s (amortized, batch size 6). 63 / 300 completed.
...
23/04/13 12:51:28 | INFO | root | Predicted structure for YP_003987266.1 with length 248, pLDDT 40.9, pTM 0.373 in 2.9s (amortized, batch size 4). 135 / 300 completed.
...
23/04/13 12:55:54 | INFO | root | Predicted structure for YP_003987173.1 with length 352, pLDDT 42.5, pTM 0.322 in 6.6s (amortized, batch size 2). 191 / 300 completed.
...
23/04/13 13:01:07 | INFO | root | Predicted structure for YP_003987272.1 with length 449, pLDDT 55.0, pTM 0.310 in 12.1s (amortized, batch size 2). 224 / 300 completed.
...
23/04/13 13:11:49 | INFO | root | Predicted structure for YP_003987159.1 with length 550, pLDDT 92.5, pTM 0.962 in 20.9s. 263 / 300 completed.
...
23/04/13 13:19:57 | INFO | root | Predicted structure for YP_003987287.1 with length 650, pLDDT 52.9, pTM 0.419 in 32.8s. 282 / 300 completed.
...
23/04/13 13:24:23 | INFO | root | Predicted structure for YP_003987282.1 with length 751, pLDDT 50.8, pTM 0.397 in 44.7s. 289 / 300 completed.
...
23/04/13 13:30:22 | INFO | root | Predicted structure for YP_003987343.1 with length 990, pLDDT 58.2, pTM 0.387 in 94.3s. 294 / 300 completed.
23/04/13 13:30:23 | INFO | root | Failed (CUDA out of memory) on sequence YP_003987327.1 of length 1297.
23/04/13 13:30:24 | INFO | root | Failed (CUDA out of memory) on sequence YP_003987190.1 of length 1387.
23/04/13 13:30:27 | INFO | root | Failed (CUDA out of memory) on sequence YP_003987364.1 of length 1624.
23/04/13 13:30:27 | INFO | root | Failed (CUDA out of memory) on sequence YP_003987350.1 of length 1651.
23/04/13 13:30:28 | INFO | root | Failed (CUDA out of memory) on sequence YP_003987358.1 of length 1657.
23/04/13 13:30:28 | INFO | root | Failed (CUDA out of memory) on sequence YP_003987191.1 of length 1937.
As we can see above esm-fold batches shorter sequences. The default number
of tokens (aka residues) per batch is 1024 for version 2.0.0. If memory
problems occur with batches of shorter sequences try reducing
--max-tokens-per-batch. Because of the batching mechanism GPU
memory usage did not increase consistently during the run above. Below is a
graph of GPU memory usage during the run above. Some batch size transitions are
highlighted.
esm-fold run with default settings on 300 mimivirus
proteins. Batch sizes (n) are indicated at the top.In our case failures occurred with the longest sequences running
individually with the largest model topping out at 990 aa. Let's see the effect
of chunking attention computation with --chunk-size and offloading
to CPU with --cpu-offload on memory consumption for the largest 14
sequences from the test set above ranging from 702 to 1937 residues:
[user@cn3144]$ esm-fold -i test2.fa -o test2_models1 --cpu-offload [user@cn3144]$ esm-fold -i test2.fa -o test2_models2 --chunk-size 128 [user@cn3144]$ esm-fold -i test2.fa -o test2_models3 --chunk-size 64 [user@cn3144]$ esm-fold -i test2.fa -o test2_models4 --chunk-size 32

esm-fold run with different settings for
the 14 largest protein sequences from Fig. 1It is clear that --cpu-offload did not allow esm-fold to generate models
for any of the 6 sequences that failed with default settings. However, any level of chunking
consumed significantly less GPU memory for the first 8 models than default settings and
allowed esm-fold to generate models up to 1937. There is no clear difference in either
runtime or memory usage between the different chunk sizes.
It is also possible to execute python code using esm. Here is an interactive example:
[user@cn3144]$ esm-python Python 3.8.16 | packaged by conda-forge | (default, Feb 1 2023, 16:01:55) [GCC 11.3.0] on linux Type "help", "copyright", "credits" or "license" for more information. >>> import esm >>> import torch >>> model, alphabet = esm.pretrained.esm2_t33_650M_UR50D()
Note that this python install includes only the dependencies for esm-fold. No additional packages will be added.
Inverse folding - sample 3 sequence designs for the golgi casein kinase structure
[user@cn3144]$ sample_sequences.py $ESM_TEST_DATA/5YH2.pdb \
--chain C --temperature 1 --num-samples 3 --outpath sampled_sequences.fasta
Exit the sinteractive session
[user@cn3144]$ exit salloc.exe: Relinquishing job allocation 46116226 [user@biowulf]$
Create a batch input file (e.g. esm.sh), which uses the input file 'esm.in'. For example:
#!/bin/bash module load esm/2.0.0 export TMPDIR=/lscratch/$SLURM_JOB_ID [[ -d $TMPDIR ]] || exit 1 esm-fold -i $ESM_TEST_DATA/mimi_all.fa -o mimi_all --chunk-size 128
Submit this job using the Slurm sbatch command.
sbatch --partition=gpu --time=12:00:00 --cpus-per-task=8 --mem=48g --gres=lscratch:100,gpu:a100:1 esm.sh
Allocate an interactive session and run the program. Sample session:
The small esm3_sm_open_v1 model is available on biowulf via the esm python package.
Note that this tool requires allocation of at least 10GB of lscratch to use as the huggingface
cache (pre-popolated with the model from /fdb.
[user@biowulf]$ sinteractive --gres=lscratch:100,gpu:a100:1 --mem=24g --cpus-per-task=8
salloc.exe: Pending job allocation 46116226
salloc.exe: job 46116226 queued and waiting for resources
salloc.exe: job 46116226 has been allocated resources
salloc.exe: Granted job allocation 46116226
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn3144 are ready for job
[user@cn3144]$ module load esm/3.0.1
[+] Loading esm 3.0.1
[user@cn3144]$ esm-python
setting up ephemeral huggingface cache in lscratch; one second ...
Python 3.11.2 (main, May 2 2024, 11:59:08) [GCC 12.2.0] on linux
Type "help", "copyright", "credits" or "license" for more information.
>>> from esm.models.esm3 import ESM3
>>> from esm.sdk.api import ESM3InferenceClient, ESMProtein, GenerationConfig
>>> # This will load the model weights and instantiate the model from our cache
>>> model: ESM3InferenceClient = ESM3.from_pretrained("esm3_sm_open_v1").to("cuda") # or "cpu"
>>> # Generate a completion for a partial Carbonic Anhydrase (2vvb)
>>> prompt = "___________________________________________________DQATSLRILNNGHAFNVEFDDSQDKAVLKGGPLDGTYRLIQFHFHWGSLDGQGSEHTVDKKKYAAELHLVHWNTKYGDFGKAVQQPDGLAVLGIFLKVGSAKPGLQKVVDVLDSIKTKGKSADFTNFDPRGLLPESLDYWTYPGSLTTPP___________________________________________________________"
>>> protein = ESMProtein(sequence=prompt)
>>> # Generate the sequence, then the structure. This will iteratively unmask the sequence track.
>>> protein1 = model.generate(protein, GenerationConfig(track="sequence", num_steps=8, temperature=0.7))
>>> protein2 = model.generate(protein, GenerationConfig(track="sequence", num_steps=8, temperature=0.7))
>>> print(protein1)
ESMProtein(sequence='MKSFRRLLVLLAAASALGAAFACDKGKQQSPTDTQPLDRRENLNPLELSGYDQATSLRILNNGHAFNVEFDDSQDKAVLKGGPLDGTYRLIQFHFHWGSLDGQGSEHTVDKKKYAAELHLVHWNTKYGDFGKAVQQPDGLAVLGIFLKVGSAKPGLQKVVDVLDSIKTKGKSADFTNFDPRGLLPESLDYWTYPGSLTTPPCDESVIWTVFKRPVELSRAQLEKFRGLTTDDKGQPLEFNFRERTKLAGGDKRVVTERGX', secondary_structure=None, sasa=None, function_annotations=None, coordinates=None, plddt=None, ptm=None)
>>> print(protein2)
ESMProtein(sequence='MAQEFFQRDTHAIAEDYDGNHSGSKNGQHQITIGTKAAVVEPLKPLQLQGYDQATSLRILNNGHAFNVEFDDSQDKAVLKGGPLDGTYRLIQFHFHWGSLDGQGSEHTVDKKKYAAELHLVHWNTKYGDFGKAVQQPDGLAVLGIFLKVGSAKPGLQKVVDVLDSIKTKGKSADFTNFDPRGLLPESLDYWTYPGSLTTPPCYESVVWTVFKEPIEVTGEQLAKFRALLSDPEGEPLLNNFRPQQDKENVTVVRSPSAFH', secondary_structure=None, sasa=None, function_annotations=None, coordinates=None, plddt=None, ptm=None)
>>> # We can show the predicted structure for the generated sequence.
>>> protein1 = model.generate(protein1, GenerationConfig(track="structure", num_steps=8))
>>> protein1.to_pdb("./generation.pdb")
>>> # Then we can do a round trip design by inverse folding the sequence and recomputing the structure
>>> protein1.sequence = None
>>> protein1 = model.generate(protein1, GenerationConfig(track="sequence", num_steps=8))
>>> protein1.coordinates = None
>>> protein1 = model.generate(protein1, GenerationConfig(track="structure", num_steps=8))
>>> protein1.to_pdb("./round_tripped.pdb")
>>> # function annotation
>>> protein3 = model.generate(protein1, GenerationConfig(track="function"))
>>> protein2.function_annotations[0]
FunctionAnnotation(label='carbonic', start=27, end=254)
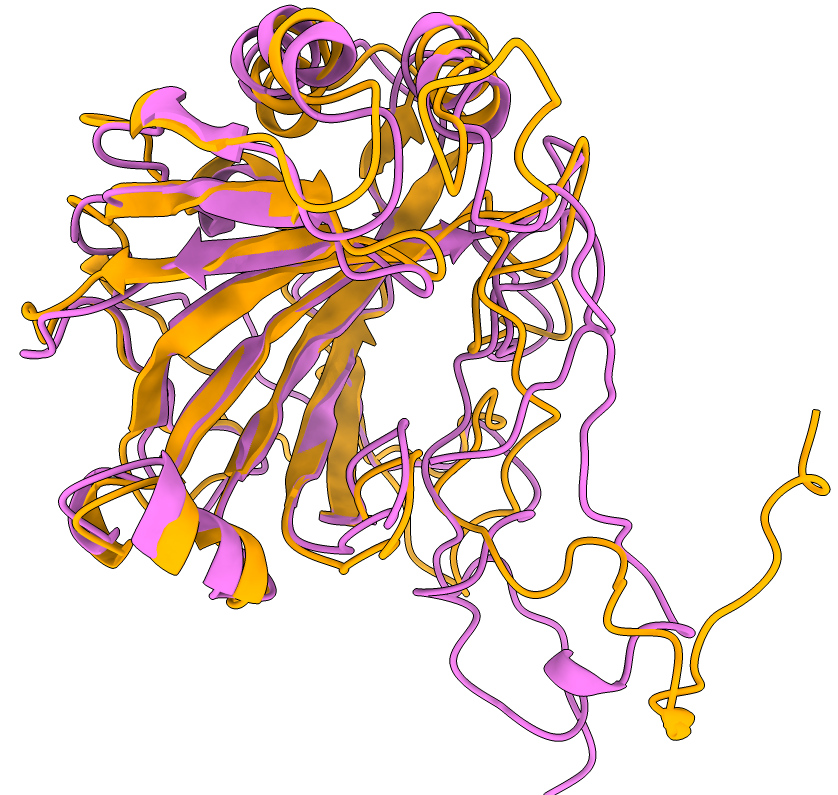
Version 3.1.2 also includes the esmc_300m and esmc_600m models.
We also provide a utility script to install a jupyter kernel so you can use ESM3 in jupyter notebooks:
[user@biowulf]$ sinteractive --gres=lscratch:20 salloc.exe: Pending job allocation 46116226 salloc.exe: job 46116226 queued and waiting for resources salloc.exe: job 46116226 has been allocated resources salloc.exe: Granted job allocation 46116226 salloc.exe: Waiting for resource configuration salloc.exe: Nodes cn3144 are ready for job [user@cn3144]$ module load esm/3.0.1 [+] Loading esm 3.0.1 # only has to be done once for a particular version [user@cn3144]$ esm-install-kernel
After installing the kernel it will be available when you start a new jupyter instance (with the required resources).