The GeoMx NGS Pipeline, developed by NanoString, is an essential part of the GeoMx NGS workflow. The Pipeline processes RNA-sequencing files (FASTQ files) from Illumina sequencers according to parameters defined in the Configuration File (which is generated from the GeoMx DSP run). The Pipeline processes information from these files and outputs .dcc files, which can then be uploaded to the GeoMx DSP system for data analysis.
Allocate an interactive session and run the program.
Sample session (user input in bold):
[user@biowulf ~]$ sinteractive -c8 --gres=lscratch:60 --mem=16g
salloc.exe: Pending job allocation 13797412
salloc.exe: job 13797412 queued and waiting for resources
salloc.exe: job 13797412 has been allocated resources
salloc.exe: Granted job allocation 13797412
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn0933 are ready for job
[user@cn0933 ~]$ cd /lscratch/$SLURM_JOB_ID
[user@cn0933 13797412]$ module load geomx_ngs_pipeline
[+] Loading geomx_ngs_pipeline 2.0.0.16 on cn0933
[+] Loading singularity 3.7.3 on cn0933
[user@cn0933 13797412]$ cp -r $GEOMX_NGS_PIPELINE_TESTDATA .
[user@cn0933 13797412]$ cd TESTDATA
[user@cn0933 TESTDATA]$ geomxngspipeline --in=FASTQ --out=test \
--ini=3sampleAOIs_20201119_GeoMxNGSPipelinev2.ini \
--threads=$SLURM_CPUS_PER_TASK
2021-04-29 18:30:35.9155 Info args: "--in=FASTQ", "--out=test", "--ini=3sampleAOIs_20201119_GeoMxNGSPipelinev2.ini", "--threads=8"
2021-04-29 18:30:36.0062 Info Build version: "2.0.0.16"
2021-04-29 18:30:36.0080 Info RAM available: 378. CPU count: 8
2021-04-29 18:30:36.0101 Info Parsing processing settings from ini file...
2021-04-29 18:30:36.0483 Info Ini parsed successfully.
2021-04-29 18:30:36.0858 Info Processing FASTQ with next params: Input dir path: FASTQ
Outpur dir path: test
.ini file path: 3sampleAOIs_20201119_GeoMxNGSPipelinev2.ini
Project name:
Quality trim score: 20
2color quality trimming: False
Adapter 1:
Adapter 2:
Adapter trim match length: 10
Adapter trim max mismatch: 3
Stitching max mismatch: 2
Stitch shift: True
Skip stitching: False
Barcode max mismatch: 1
Dedup HD: 1
Dedup reads limit: 2000000000
Parallel threads count: 8
Save interim files: False
Translation file:
Single FASTQ processing mode: False
Use single read if stitching failed: False
Mode: Process
2021-04-29 18:30:36.1159 Info Processing sample: DSP-1001660005876-A12
2021-04-29 18:30:36.1159 Info Processing sample: DSP-1001660005876-C01
[...snip]
2021-04-29 18:35:38.7822 Info All done in 302695 ms
[user@cn0933 TESTDATA]$ ls -l test/
total 320
-rw-r--r-- 1 user user 79161 Apr 29 18:35 DCC-20210429.zip
-rw-r--r-- 1 user user 682 Apr 29 18:30 DSP-1001660005876-A01.dcc
-rw-r--r-- 1 user user 648 Apr 29 18:30 DSP-1001660005876-A01.stats
-rw-r--r-- 1 user user 109764 Apr 29 18:35 DSP-1001660005876-A12.dcc
-rw-r--r-- 1 user user 730 Apr 29 18:35 DSP-1001660005876-A12.stats
-rw-r--r-- 1 user user 109171 Apr 29 18:35 DSP-1001660005876-C01.dcc
-rw-r--r-- 1 user user 728 Apr 29 18:35 DSP-1001660005876-C01.stats
-rw-r--r-- 1 user user 3550 Apr 29 18:35 processing.log
-rw-r--r-- 1 user user 277 Apr 29 18:35 summary.txt
[user@cn0933 TESTDATA]$ exit
exit
salloc.exe: Relinquishing job allocation 13797412
salloc.exe: Job allocation 13797412 has been revoked.
[user@biowulf ~]$
It's also possible to interact with the GeoMX NGS Pipeline software by starting a server, creating an ssh tunnel, and connecting with the GeoMX client. You can download the GUI client here.
First, establish an interactive session with an ssh tunnel, and start the GeoMX NGS Pipeline server.
[user@biowulf ~]$ sinteractive -c4 --gres=lscratch:10 --mem=8g --tunnel
salloc.exe: Pending job allocation 14389345
salloc.exe: job 14389345 queued and waiting for resources
salloc.exe: job 14389345 has been allocated resources
salloc.exe: Granted job allocation 14389345
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn0881 are ready for job
srun: error: x11: no local DISPLAY defined, skipping
error: unable to open file /tmp/slurm-spank-x11.14389345.0
slurmstepd: error: x11: unable to read DISPLAY value
Created 1 generic SSH tunnel(s) from this compute node to
biowulf for your use at port numbers defined
in the $PORTn ($PORT1, ...) environment variables.
Please create a SSH tunnel from your workstation to these ports on biowulf.
On Linux/MacOS, open a terminal and run:
ssh -L 40075:localhost:40075 user@biowulf.nih.gov
For Windows instructions, see https://hpc.nih.gov/docs/tunneling
[user@cn0881 ~]$ module load geomx_ngs_pipeline
[+] Loading geomx_ngs_pipeline 2.0.0.16 on cn0881
[+] Loading singularity 3.7.3 on cn0881
[user@cn0881 ~]$ GeoMxNGSPipeline_API
info: Microsoft.Hosting.Lifetime[0]
Now listening on: http://0.0.0.0:40075
info: Microsoft.Hosting.Lifetime[0]
Application started. Press Ctrl+C to shut down.
info: Microsoft.Hosting.Lifetime[0]
Hosting environment: Production
info: Microsoft.Hosting.Lifetime[0]
Content root path: /var/GeoMxNGSPipeline
Pay attention to the port number where your server was started. In the example above it was 40075. In a new terminal or powershell window, use the port number to estabish an ssh tunnel between your local computer and the Biowulf login node.
Windows PowerShell
Copyright (C) Microsoft Corporation. All rights reserved.
Try the new cross-platform PowerShell https://aka.ms/pscore6
PS C:\Users\User> ssh -L 40075:localhost:40075 user@biowulf.nih.gov
***WARNING***
You are accessing a U.S. Government information system, which includes
(1) this computer, (2) this computer network, (3) all computers
connected to this network, and (4) all devices and storage media
attached to this network or to a computer on this network. This
information system is provided for U.S. Government-authorized use only.
Unauthorized or improper use of this system may result in disciplinary
action, as well as civil and criminal penalties.
By using this information system, you understand and consent to the
following:
* You have no reasonable expectation of privacy regarding any
communications or data transiting or stored on this information system.
At any time, and for any lawful Government purpose, the government may
monitor, intercept, record, and search and seize any communication or
data transiting or stored on this information system.
* Any communication or data transiting or stored on this information
system may be disclosed or used for any lawful Government purpose.
--
Notice to users: This system is rebooted for patches and maintenance on
the first Monday of every month at 7:15AM unless Monday is a holiday, in
which case it is rebooted the following Tuesday. Running cluster jobs
are not affected by the monthly reboot.
user@biowulf.nih.gov's password:
Last login: Wed May 5 13:19:20 2021 from cit01234567.cit.nih.gov
[user@biowulf ~]$
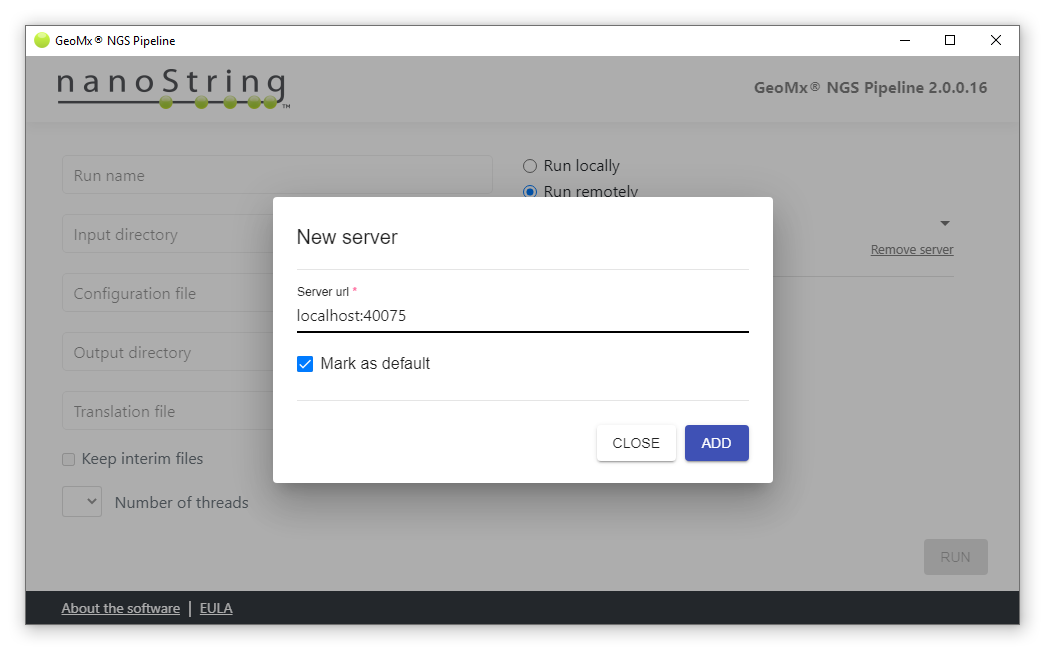
Now you can connect your GeoNX NGS Pipeline client to the server using the address localhost:<port number> where <port number> is the the same number you used to establish the tunnel.
The file browser feature only allows you to see files in your home directory. You can create symlinks in your home directory so that you can browse files in other locations. For instance, if you want to analyze data in your /data/$USER directory, you can create a symlink like so:
[user@biowulf ~]$ cd ~ [user@biowulf ~]$ ln -s /data/$USER data
Create a batch input file (e.g. geomx_ngs_pipeline.sh). For example:
#!/bin/bash
set -e
module load geomx_ngs_pipeline
cd /data/$USER/location/of/data
geomxngspipeline --in=datadir --out=outdir \
--ini=sample_GeoMxNGSPipelinev2.ini \
--threads=$SLURM_CPUS_PER_TASK
Submit this job using the Slurm sbatch command.
sbatch [--cpus-per-task=#] [--mem=#] geonx_ngs_pipeline.sh
Create a swarmfile (e.g. geomx_ngs_pipeline.swarm). For example:
geomxngspipeline --in=datadir1 --out=outdir1 --ini=sample1.ini --threads=$SLURM_CPUS_PER_TASK geomxngspipeline --in=datadir2 --out=outdir2 --ini=sample2.ini --threads=$SLURM_CPUS_PER_TASK geomxngspipeline --in=datadir3 --out=outdir3 --ini=sample3.ini --threads=$SLURM_CPUS_PER_TASK geomxngspipeline --in=datadir4 --out=outdir4 --ini=sample4.ini --threads=$SLURM_CPUS_PER_TASK
Submit this job using the swarm command.
swarm -f geomx_ngs_pipeline.swarm [-g #] [-t #] --module geomx_ngs_pipelinewhere
| -g # | Number of Gigabytes of memory required for each process (1 line in the swarm command file) |
| -t # | Number of threads/CPUs required for each process (1 line in the swarm command file). |
| --module geomx_ngs_pipeline | Loads the geomx_ngs_pipeline module for each subjob in the swarm |