Please read Making Effective Use of Biowulf's multinode partition before running multinode Gromacs jobs.
For information about Gromacs performance, see the following papers:
Dec 2018
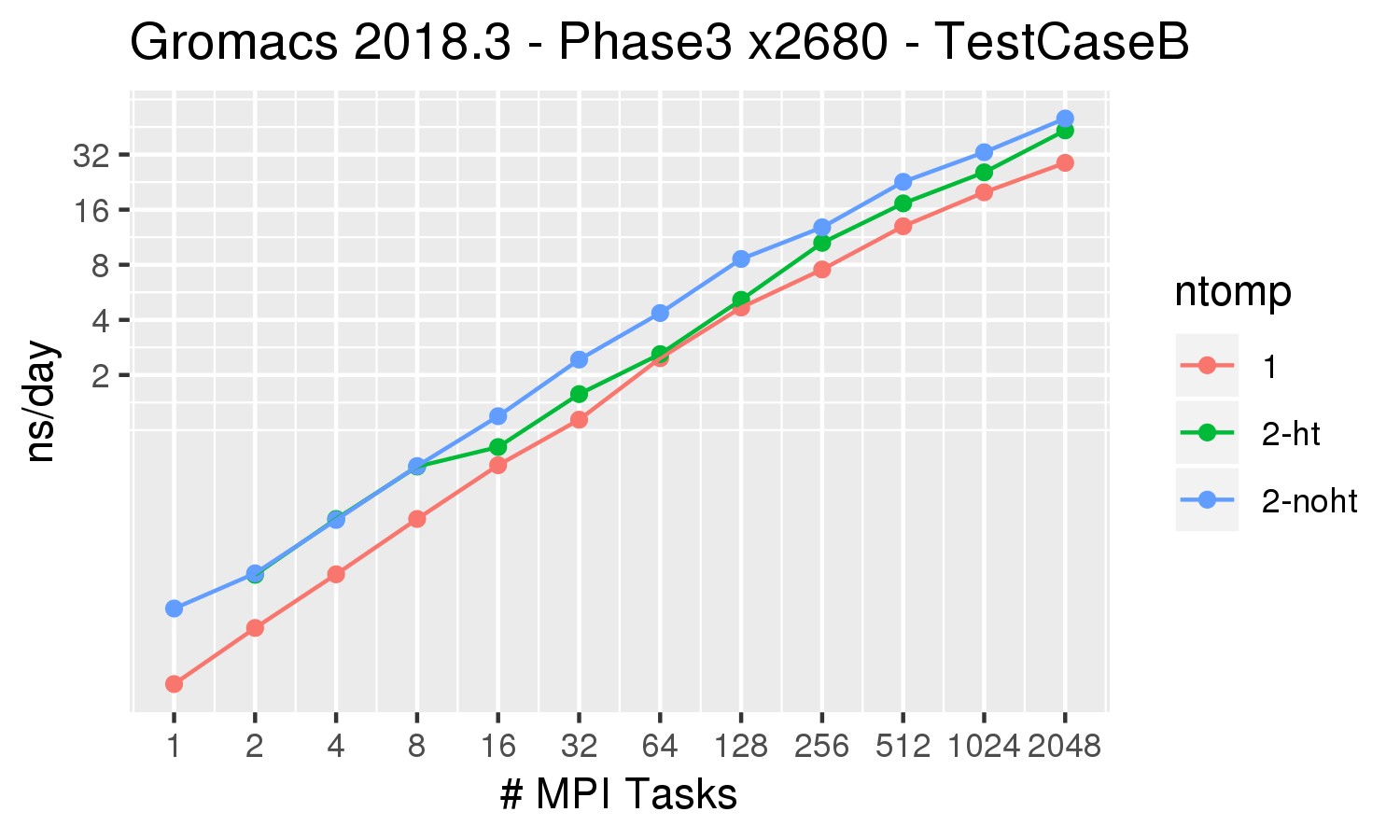
Gromacs Test Case B from the Unified European Applications Benchmark Suite.
Description from their documentation: A model of cellulose and
lignocellulosic biomass in an aqueous solution. This system of 3.3M atoms is inhomogeneous, at least with GROMACS 4.5. This system uses
reaction-field electrostatics instead of PME and therefore should scale well.
Gromacs 2018.3 was built with cmake 3.4.3, Intel 2018.1.163 compiler, OpenMPI 2.1.2 All runs were performed on the Biowulf Phase3 nodes, which are 28 x 2.4 GHz (Intel E5-2680v4), hyperthreading enabled, connected via 56 Gb/s FDR Infiniband (1.11:1).
| ntomp = 1 | orange line | 1 thread per MPI task, 1 MPI task per physical core. |
| ntomp = 2-ht | green line | 2 threads per MPI task, hyperthreading enabled. This effectively uses the same number of cores as ntomp=1, but uses both hyperthreaded CPUs on each core |
| ntomp = 2-noht | blue line | 2 threads per MPI task, no hyperthreading. This uses twice as many cores as the options above. |
For this benchmark, for small numbers of MPI tasks it is worth using 2 threads per task, and utilizing the hyperthreading on the cores. For larger numbers of tasks, the all-MPI (ntomp =1 ) gives the best performance.
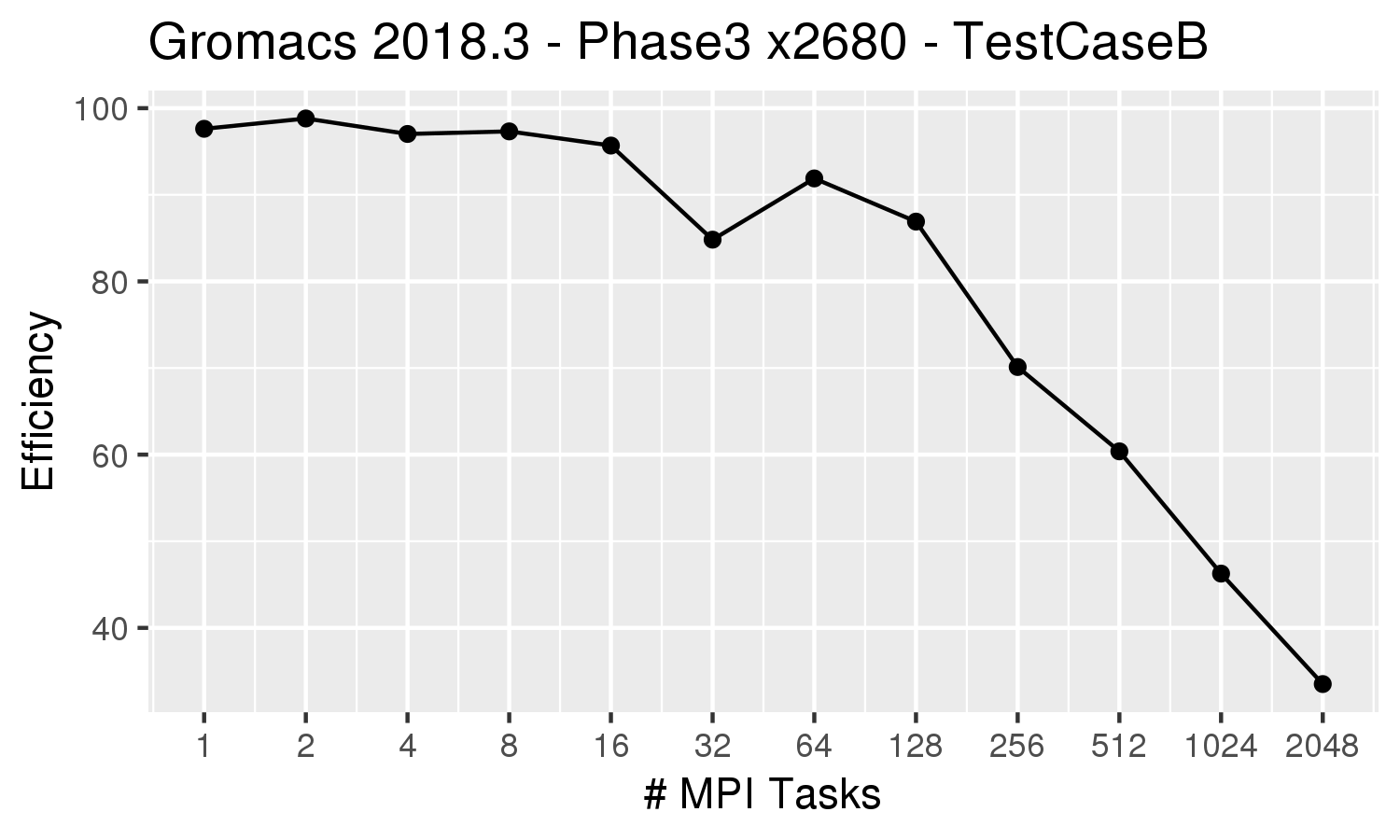
| The efficiency plot on the left for the all-MPI (ntomp=1) runs indicate that the efficiency drops below 70% for more than 256 MPI tasks. We recommend that multinode jobs maintain at least 70% efficiency. If running on the Phase 3 x2680 nodes, which have 28 cores per node,
it would therefore be best to run on 9 nodes (252 MPI processes). An appropriate submission command would be:
sbatch --partition=multinode \ --constraint=x2680 --ntasks=252 \ --ntasks-per-core=1 --time=4-00:00:00 --exclusive jobscript |
May 2017
Gromacs Test Case A from the Unified European Applications Benchmark Suite.
Description from their documentation: The ion channel system is the membrane protein GluCl,
which is a pentameric chloride channel embedded in a lipid bilayer. This system contains roughly 150,000 atoms, and is a quite challenging parallelization case due to the small size. However, it is likely one
of the most wanted target sizes for biomolecular simulations due to the importance of these
proteins for pharmaceutical applications. It is particularly challenging due to a highly inhomogeneous and anisotropic environment in
the membrane, which poses hard challenges for load balancing with domain decomposition.
Gromacs 2018.3 was built with cmake 3.4.3, Intel 2018.1.163 compiler, OpenMPI 2.1.2 All runs were performed on the Biowulf Phase3 nodes, which are 28 x 2.4 GHz (Intel E5-2680v4), hyperthreading enabled, connected via 56 Gb/s FDR Infiniband (1.11:1).
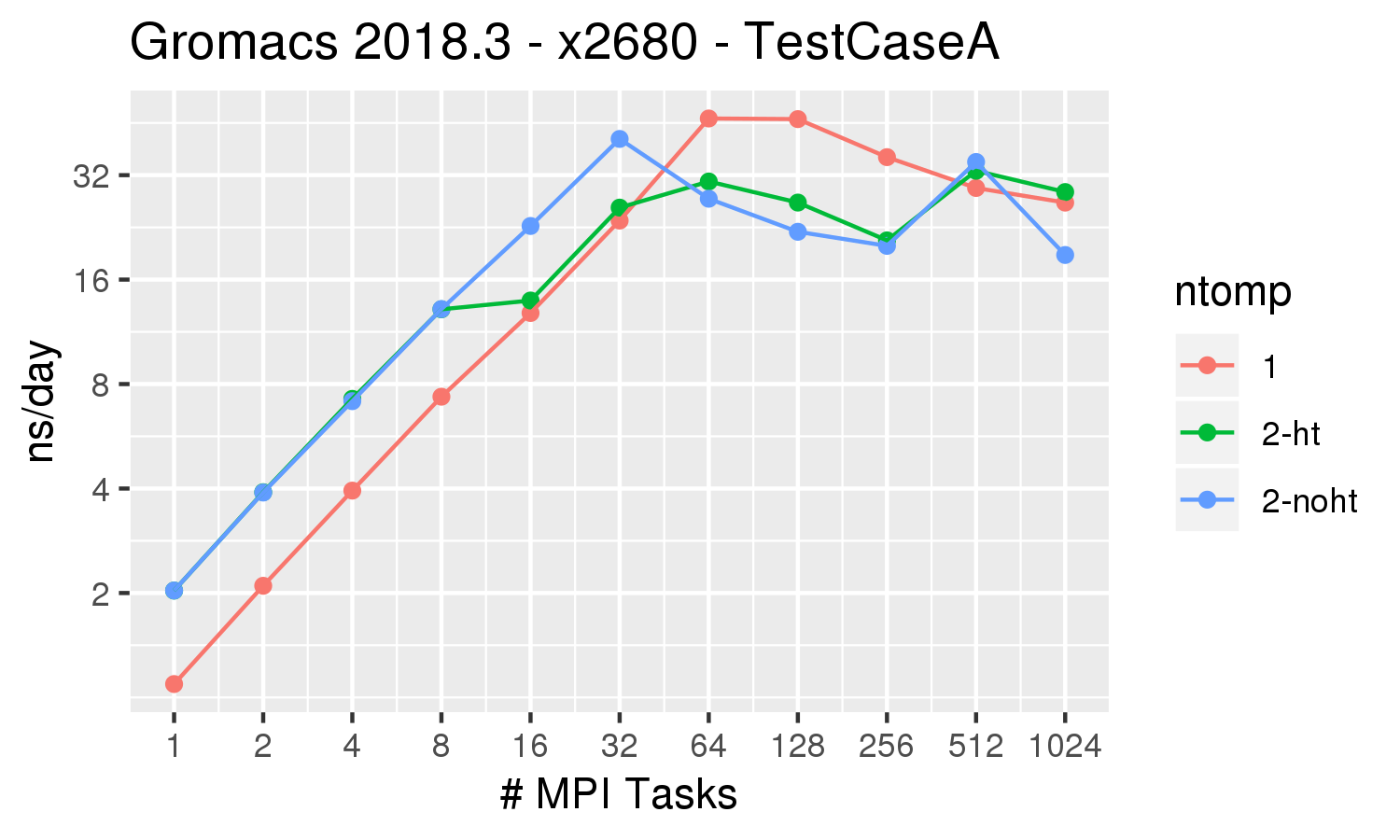
| ntomp = 1 | orange line | 1 thread per MPI task, 1 MPI task per physical core. |
| ntomp = 2-ht | green line | 2 threads per MPI task, hyperthreading enabled. This effectively uses the same number of cores as ntomp=1, but uses both hyperthreaded CPUs on each core |
| ntomp = 2-noht | blue line | 2 threads per MPI task, no hyperthreading. This uses twice as many cores as the options above. |
For this benchmark, the best performance is obtained by running 64 MPI tasks with 1 thread. The performance drops signficantly beyond 128 tasks (cores) for this relatively small molecular simulation.
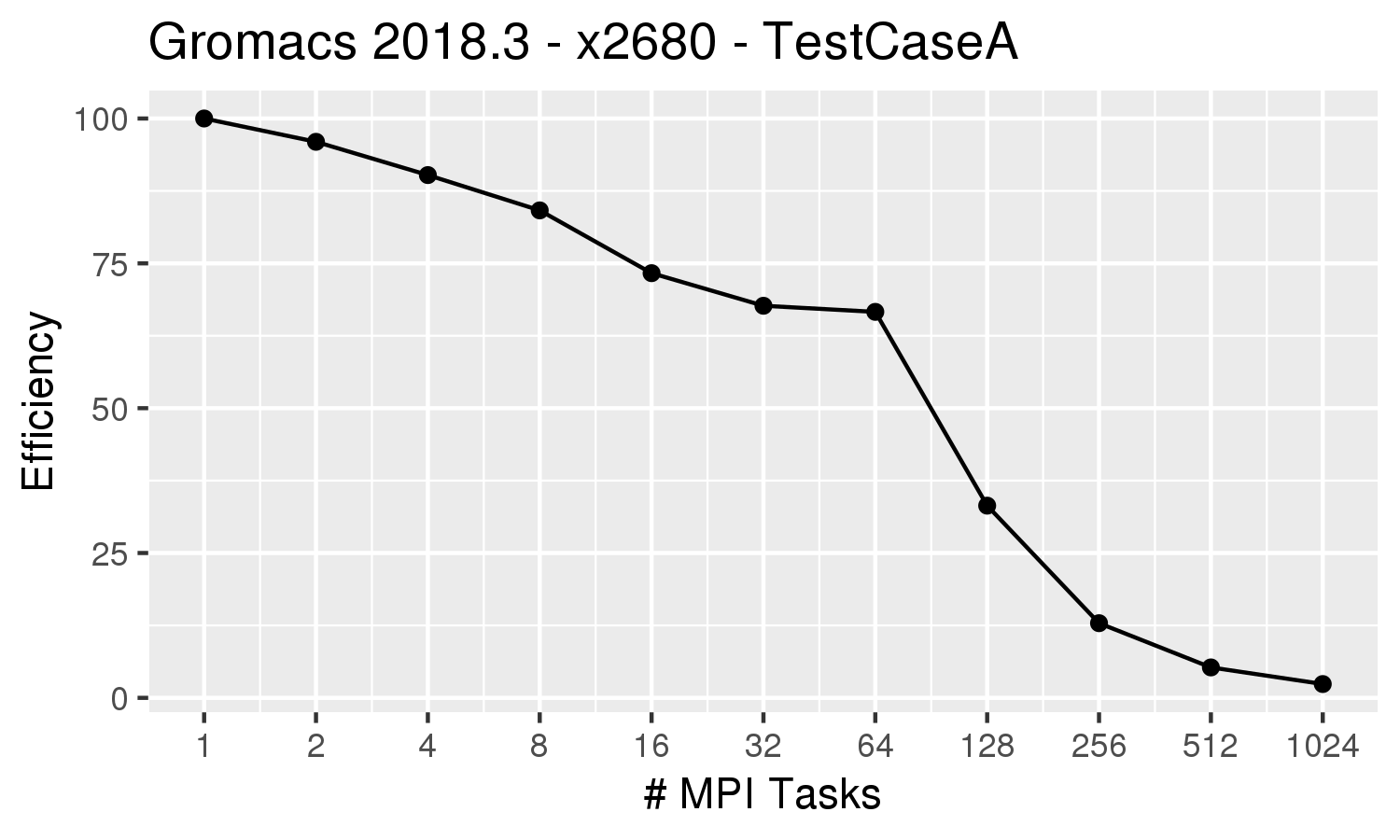
| The efficiency plot on the left for the all-MPI (ntomp=1) runs indicate that the efficiency drops below 70% for more than 16 MPI tasks. If running on the Phase 3 x2680 nodes,
which have 28 cores per node,
it would therefore be best to utilize 1 full node (28 MPI processes). Since this is a single-node job, it could be run in the 'norm' partition. An appropriate submission command for this job would be:
sbatch --partition=norm \ --constraint=x2680 --ntasks=28 \ --ntasks-per-core=1 --time=4-00:00:00 --exclusive jobscript |
The following benchmark uses the adh_cubic job from the Gromacs GPU documentation. Please read the discussion on that page about GPU acceleration.
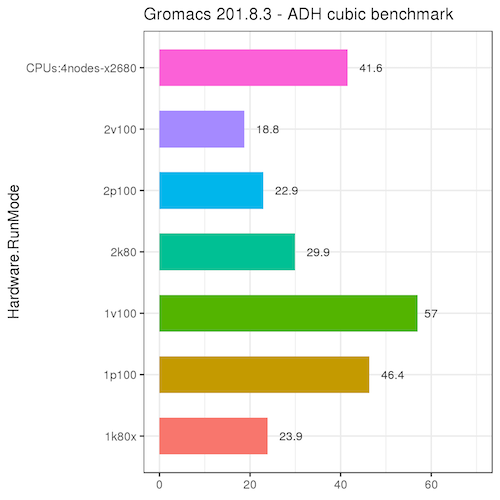
| For this benchmark, there is a significant advantage to running Gromacs on the GPUs, especially the p100 and v100 nodes, over IB nodes. There is typically no advantage to running on more than a single GPU device.
Single-GPU jobs were run on a single GPU with ntasks=1 and ntomp=7. (Each GPU node has 4 GPU devices and 56 CPUs, so the jobs were set up to utilize 1 GPU and 1/4 of the CPUs). 2-GPU jobs were run with ntasks=2 and ntomp=7. These results may be different for larger molecular systems, so please run your own benchmarks to verify that your jobs benefit from the GPUs (and send us any interesting results!)
|