The multinode partition on Biowulf is intended to be used for large-scale parallel jobs. There are a large amount of resources available in the form of InfiniBand-connected nodes. The per-user CPU limit for the multinode partition can be seen by typing 'batchlim'. In addition to this limit, additional CPUs can be utilized for jobs with a time limit of less than 8 hrs by submitting with the 'turbo' QOS.
However, just because you can use a large number of resources does not mean you should. Many different types of jobs actually perform more efficiently when fewer CPUs are used. In other words, adding CPUs often yields diminishing marginal returns. For example, if you double the number of CPUs you use, but the runtime of your job is only shortened by 30%, that is not a good use of resources. In general, the NIH HPC staff will ask to see proof that jobs requesting more than 512 CPUs are actually able to take advantage of them.
This page will provide information that can help you determine whether your multinode jobs are running efficiently. It also provides a few tips for more effective use of the multinode partition.
Before submitting any parallel job, you should...
Please note that much of the advice given here is based on staff experience with large-scale MPI jobs (e.g. molecular dynamics). However, the general principles hold for other types of large parallel jobs as well.
The multinode partition actually consists of three different types of nodes, as shown by freen:
biowulf% freen ... multinode 65/466 3640/26096 28 56 248g 400g cpu56,core28,g256,ssd400,x2695,ibfdr multinode 4/190 128/6080 16 32 60g 800g cpu32,core16,g64,ssd800,x2650,ibfdr multinode 312/539 17646/30184 28 56 250g 800g cpu56,core28,g256,ssd800,x2680,ibfdr ...
As shown above, the three types of nodes are older 16-core nodes with x2650 processors, newer 28-core nodes with 2.3 GHz x2695 processors, and the newest nodes with 2.4 GHz x2680 processor (despite the lower number, these processors are newer). A single parallel job should only use one type of node. In other words, a single multinode job should use all x2650, all x2695, or all x2680 nodes. Mixing a single job between node types will cause it to run in a very inefficient manner, since faster processors have to wait for slower ones.
Therefore, either "--constraint=x2695" or "--constraint=x2650" or "--constraint=x2680"
should always be used when submitting any multinode job. e.g.:
sbatch --partition=multinode --constraint=x2650 --ntasks=64 --ntasks-per-core=1 --time=168:00:00 --exclusive jobscriptwould submit a 64-core job to the x2650 nodes on the multinode partition.
Also, when using the "--exclusive" flag, please select a number of tasks ("--ntasks") that is an even multiple of the number of CPUs (or cores, if using "--ntasks-per-core=1") so that resources on nodes are not wasted.
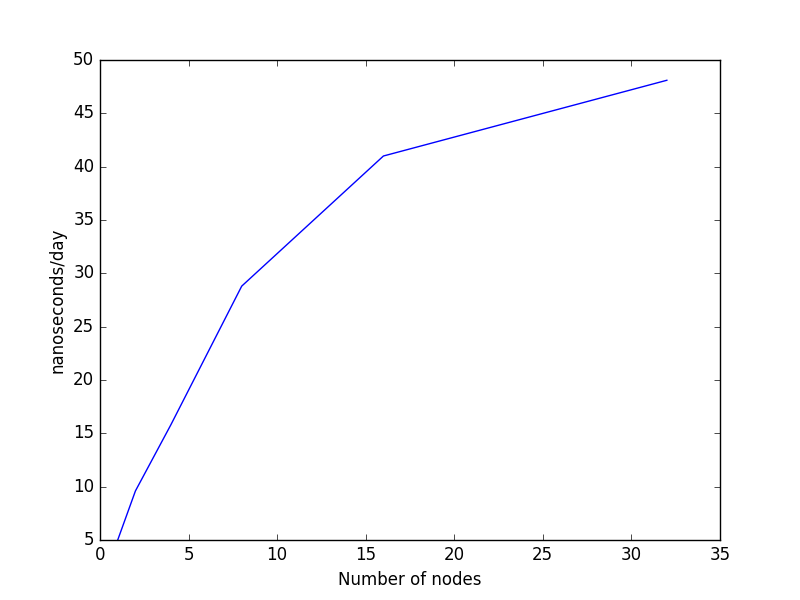
The only reliable way to see if a job will scale efficiently is to benchmark it. Benchmarking a job means running a short, representative test job multiple times at different numbers of CPUs to find an optimal point. Take as an example a molecular dynamics job. You can run short (10,000-20,000 step) benchmark jobs at different number of CPUs (e.g. using 1 28 core node, 2 28 core nodes, 4 28 core nodes, etc.) and make a table with the (extrapolated) nanoseconds per day of each job. This table might look like (for jobs run on the older, core16 nodes):
| Number of nodes | Number of cores | Nanoseconds per day |
|---|---|---|
| 1 | 16 | 5.0 |
| 2 | 32 | 9.6 |
| 4 | 64 | 15.8 |
| 8 | 128 | 28.8 |
| 16 | 256 | 41.0 |
| 32 | 512 | 48.1 |
From this data, you can calculate parallel efficiency. This is defined as:
efficiency = (work done for N CPUs)
-----------------------
(N * work for 1 CPU)
Since benchmarking on a
single core can often be time consuming and scaling within a node is generally very
good, it is sufficient for Biowulf's purposes to do this calculation on a per
node, rather than a per CPU basis. Applying this formula to the above sample scaling
data, we arrive at the following efficiencies:
| Number of nodes | Number of cores | Nanoseconds per day | Parallel efficiency |
|---|---|---|---|
| 1 | 16 | 5.0 | 1.00 (probably not really, but close enough) |
| 2 | 32 | 9.6 | 0.96 |
| 4 | 64 | 15.8 | 0.79 |
| 8 | 128 | 28.8 | 0.72 |
| 16 | 256 | 41.0 | 0.51 |
| 32 | 512 | 48.1 | 0.30 |
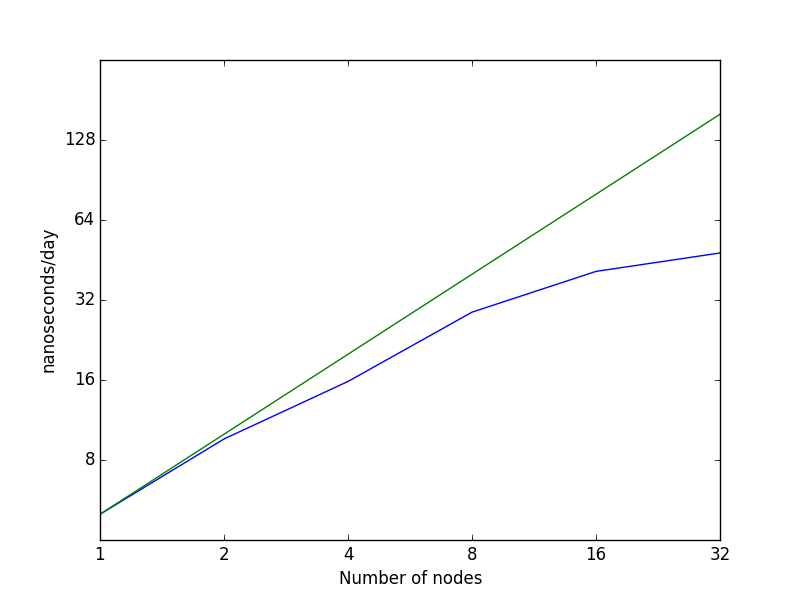
From this, we see that after 128 cores, the benefits for requesting more resources become very marginal. Therefore, for this example, the production simulation should be run at 128 cores (256 CPUs). Using more resources than this will result in an unacceptable level of waste. As a general rule of thumb, jobs should run with a number of cores where the parallel efficiency is above 0.7.
The two graphs below show the data visually. In the left hand graph, you can see how the curve becomes notably flatter beyond 8 nodes (128 cores). The right hand graph plots the data on a log-log scale (using log base 2). In this case, the ideal scaling is represented by the straight green line while actual scaling is denoted by the blue line. You can see how above 8 nodes, actual scaling deviates signficantly from ideal scaling.
Please note: it is in your own best interest to run with a number of cores that has a parallel efficiency above 0.7, since job priority is based on the amount of past CPU usage over the last few months. If you are using CPU time unproductively, your are lowering the priority of future jobs you submit, meaning that they will sit in the queue longer when the system is busy. However, you are not getting much benefit from these CPU hours. In addition, you are denying resources to other users who could use them more productively.
Most multinode jobs on Biowulf are very CPU intensive but do not require a lot of memory or perform a lot of data input and output (I/O). Again, molecular dynamics simulation jobs are archtypical in that their memory and I/O requirements are usually quite low. However, there are some multinode jobs that use large amounts of memory and I/O resources. There are special considerations for these jobs.
As with all jobs, you should estimate the memory and I/O requirements of your jobs before submitting large jobs.
To estimate the memory usage, use 'jobhist [jobnumber]' on the benchmarks you run, which will report the memory used. If your jobs are memory-intensive (require more than 2 GB/cpu), you should request enough memory but not overallocate. If you over-allocate memory or walltime, the job may remain queued for longer because the scheduler needs to find enough resources for this job. Example:
biowulf$ jobhist 25363410 JobId : 25363410 User : user Submitted : 20161025 17:16:08 Ended : 20161026 08:52:07 Submission Path : /spin1/users/user/charmm/jobs/1505-bakeoff/groel Submission Command : sbatch run-mva2-c41b1.sh Partition State Nodes CPUs Walltime Runtime MemReq MemUsed Nodelist multinode COMPLETED 16 512 01:00:00 00:04:52 1.0GB/cpu 0.0GB cn[0417,0431-0432,0458-0460,0495-0497,0525-0529,0531-0532]As you can see, this job used very little memory (< 100 MB per CPU, which is recorded as 0.0 GB due to numeric precision). If, however, the memory usage was close to the limit, you would need to consider increasing --mem-per-cpu if submitting a larger job.
You can monitor long running jobs via jobload, e.g.:
biowulf$ jobload -j 27315864
27315864 2-02:35:30 / 10-00:00:00 cn1166 56 15 27% 20.6 / 252.1 GB
2-02:35:30 / 10-00:00:00 cn1271 56 15 27% 20.6 / 252.1 GB
2-02:35:30 / 10-00:00:00 cn1272 56 15 27% 21.3 / 252.1 GB
2-02:35:30 / 10-00:00:00 cn1273 56 15 27% 20.9 / 252.1 GB
2-02:35:30 / 10-00:00:00 cn1274 56 15 27% 21.0 / 252.1 GB
2-02:35:30 / 10-00:00:00 cn1361 56 14 25% 19.4 / 252.1 GB
2-02:35:30 / 10-00:00:00 cn1362 56 14 25% 19.8 / 252.1 GB
2-02:35:30 / 10-00:00:00 cn1363 56 14 25% 19.9 / 252.1 GB
2-02:35:30 / 10-00:00:00 cn1364 56 14 25% 20.4 / 252.1 GB
2-02:35:30 / 10-00:00:00 cn1365 56 14 25% 19.7 / 252.1 GB
2-02:35:30 / 10-00:00:00 cn1366 56 14 25% 19.8 / 252.1 GB
2-02:35:30 / 10-00:00:00 cn1507 56 14 25% 19.3 / 252.1 GB
2-02:35:30 / 10-00:00:00 cn1508 56 14 25% 19.7 / 252.1 GB
2-02:35:30 / 10-00:00:00 cn1509 56 14 25% 19.6 / 252.1 GB
2-02:35:30 / 10-00:00:00 cn1510 56 14 25% 19.8 / 252.1 GB
2-02:35:30 / 10-00:00:00 cn1511 56 14 25% 19.3 / 252.1 GB
2-02:35:30 / 10-00:00:00 cn1512 56 14 25% 19.2 / 252.1 GB
This job is using much less memory than it allocated.
To estimate the I/O, sum up the amount of data to be read or written, including temporary files, divided by the runtime of the job. If all the input files are read at the beginning of the job and the output is only written at the end of the job, it is likely that the job is only briefly I/O-intensive. If the job is continuously reading and writing, it is likely to be I/O-intensive. In that case, you will need to make sure that the job does not oversaturate the storage systems. If each task can read and write data independently of all other tasks (i.e. there is no need for shared files), then using lscratch is a good option. If this is not the case, please start with small (a few node) jobs and monitor their runtime. If using more nodes causes large slow-downs, it is probable that you are oversaturating the storage system.
For example, consider a parallel image processing job that will run for ten hours and needs to read in 10,000 500 MB input images and writes out 100 1 GB high resolution composited images. In addition, the job will use 10,000 temporary files, the average size of which is 10 MB. Total I/O requirements would be (10000*(500+10))+(100*1000) = 5200000 MB of data (or about 5.2 TB). Divide this number by 36,000 seconds (to reflect the estimated 10 hour runtime of the job) to give an estimated data rate of slightly above 144 MB/sec. This is towards the upper end of what the HPC storage systems can process for a single user, so it would be wise to do some careful testing before increasing the job size. If all 10,000 input images are read simultaneously, the actual data rate at the job start may be much higher.
If possible, it would be better to use lscratch for the temporary files. This would reduce the I/O requirement by 100000 MB (100 GB). Given that the temporary files are likely to be referenced throughout the job, even this minor improvement would likely result in noticeably better job performance.
The "turbo" quality of service (QOS) is designed to give higher resource limits and a slightly higher priority to short jobs (those requesting an 8 hour or smaller time limit). Using a shorter wall time will encourage turnover of jobs and makes the usage based priority scheduling system more effective. To use turbo, add "--qos=turbo" to your sbatch submission flags. This is only valid on the multinode partition!
Note that molecular dynamics codes specifically allow simulations to be restarted from a checkpoint file. Therefore, it is often beneficial to chain together multiple short simulations.
Using turbo does not free you from the responsibility of benchmarking your code as described above. However, since the resource limits are global, you can have more total processors in use even though an individual job might use fewer processors than the limit. For example, many researchers like to conduct multiple simulations of the same system under slightly different conditions or with a different initial random seed in order to improve sampling. When the turbo QOS is used, such a researcher could have more simultaneous jobs of the same size running.
To conclude, here are some general dos and don'ts for requesting resources on the multinode partition:
| Resource | Do | Don't |
|---|---|---|
| Number of CPUs | Benchmark your job as far as possible to determine a reasonable number of CPUs to run on for good parallel efficiency. | Request the maximum possible number of CPUs without knowing that your job will scale. The HPC staff will ask submitters of large jobs (more than 512 CPUs) to demonstrate scaling. In addition, larger jobs will usually wait longer in the queue before starting. |
| Wall times | Break your work down into the smallest reasonable chunk size. Request a wall time based on benchmarking that covers how long you expect the job to run for plus a 15%-25% buffer. If benchmarking cannot give an accurate wall time for a production run, run a single production job and use the amount of time it took plus a buffer as the wall time for similar jobs. | Submit all jobs with a wall time of 10 days. Jobs with longer wall times cannot be scheduled as efficiently, and thus they will wait longer in the queue when the system is busy. |
| Memory | Use memory utilization from small benchmark jobs to guide resource requests of future jobs. Use jobload and jobhist to monitor utilization. | Just guess at how much memory your job needs. Jobs that exceed their memory limits will be killed by the batch system. Conversly, requesting much more memory than needed will make it harder to schedule your job and may delay its start. |
| I/O | Start small with jobs that read and write a lot of data and scale them up gradually when you know that they are running well and your disk quota is sufficient to accommodate any new data. | Submit a large parallel job that writes lots of files without testing that the storage system will be able to absorb the I/O. |
| Other assistance | Ask the HPC staff for assistance if you are not sure what the best strategy for running your workload is, or if you have questions about how to construct a reasonable resource specification. | Plow on ahead without knowing whether what you are doing is making good use of a shared resource. |