Guppy is a basecaller from Oxford Nanopore Technologies. Current versions require GPUs to run.
Guppy is no longer supported by Oxford Nanopore. They recommend users to switch to Dorado.
--gres=gpu:1 --constraint='gpup100|gpuv100|gpuv100x|gpua100'$GUPPY_TEST_DATAAllocate an interactive session with
a suitable GPU for this example. Note that the example data is as subset of
data from a synthetic microbial community (see
Nicholls
et al) sequenced with the SQK-LSK109 1D sequencing kit in a FLO-MIN106 flowcell.
Sample session
(user input in bold):
[user@biowulf]$ sinteractive --gres=gpu:p100:1,lscratch:200 --mem=16g --cpus-per-task=6
salloc.exe: Pending job allocation 46116226
salloc.exe: job 46116226 queued and waiting for resources
salloc.exe: job 46116226 has been allocated resources
salloc.exe: Granted job allocation 46116226
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn2369 are ready for job
[user@cn2369 ~]$ cd /lscratch/$SLURM_JOB_ID
[user@cn2369 ~]$ module load guppy
[user@cn2369 ~]$ cp -rL ${GUPPY_TEST_DATA:-none}/* .
[user@cn2369 ~]$ ls -lh
drwxr-xr-x 3 user group 4.0K Sep 13 09:13 Zymo-GridION-EVEN-BB-SN
[user@cn2369 ~]$ du -sh Zymo-GridION-EVEN-BB-SN
11G Zymo-GridION-EVEN-BB-SN
[user@cn2369 ~]$ find Zymo-GridION-EVEN-BB-SN -name '*.fast5' -printf '.' | wc -c
160000
[user@cn2369 ~]$ guppy_basecaller --print_workflows | grep SQK-LSK109 | grep FLO-MIN106
FLO-MIN106 SQK-LSK109 dna_r9.4.1_450bps_hac
FLO-MIN106 SQK-LSK109-XL dna_r9.4.1_450bps_hac
[user@cn2369 ~]$ guppy_basecaller --input_path Zymo-GridION-EVEN-BB-SN --recursive \
--flowcell FLO-MIN106 --kit SQK-LSK109 \
-x cuda:all \
--records_per_fastq 0 \
--compress_fastq \
--save_path fastq
ONT Guppy basecalling software version x.x.x
config file: /opt/ont/guppy/data/dna_r9.4.1_450bps_hac.cfg
model file: /opt/ont/guppy/data/template_r9.4.1_450bps_hac.jsn
input path: Zymo-GridION-EVEN-BB-SN
save path: fastq
chunk size: 1000
chunks per runner: 512
records per file: 0
fastq compression: ON
num basecallers: 4
gpu device: cuda:all
kernel path:
runners per device: 4
Found 160000 fast5 files to process.
Init time: 4748 ms
0% 10 20 30 40 50 60 70 80 90 100%
|----|----|----|----|----|----|----|----|----|----|
***************************************************
Caller time: 1593158 ms, Samples called: 6901545514, samples/s: 4.33199e+06
Finishing up any open output files.
Basecalling completed successfully.
[user@cn2369 ~]$ exit
salloc.exe: Relinquishing job allocation 46116226
[user@biowulf ~]$
guppy appears to number the first available GPU as GPU 0 even if it is in fact not the
first GPU (i.e. CUDA_VISIBLE_DEVICES=0). The way to use all allocated GPUs is to
use -x cuda:all.
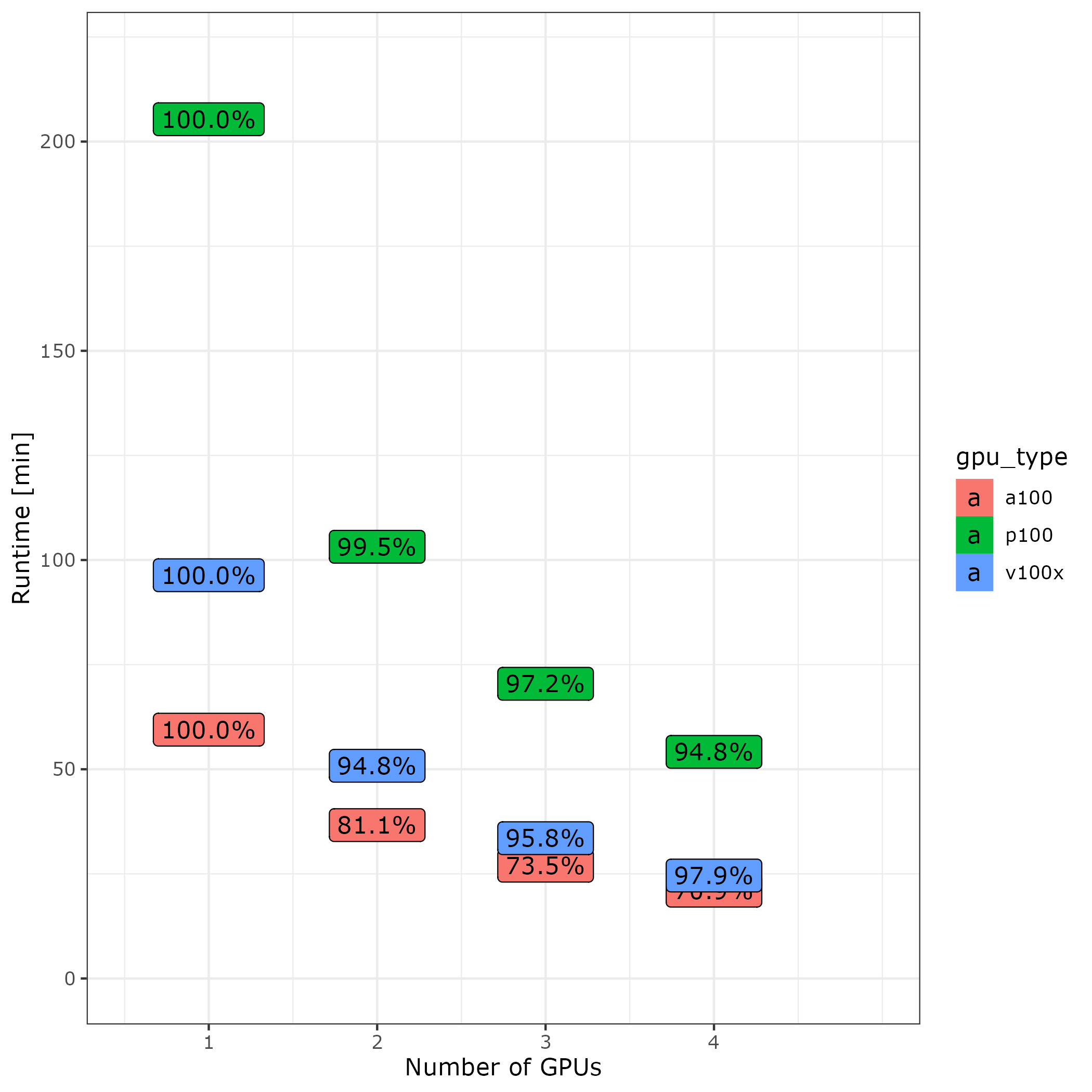
For a larger example data set, guppy_basecaller (6.4.2) runs 2.1x faster on a single V100x than a P100 and 1.6x faster on a single A100 than on a V100x for an overall speedup of 3.4 between P100 and A100. The graph below shows runtime and efficiency for 1-4 GPUs of each type using all default settings, 6CPUs/GPU, and 20GB of memory / GPU without compressing output. Input and output were located in lscratch.
When scaling CPUs with GPUs guppy 6.4.2 performs efficiently with multiple V100x and P100 GPUs. However no more than 2 A100 should be used.
Note also that the default options are not alsways optimal for all GPUs. For
example using --chunks_per_runner 1024 --chunk_size 4000 in the
benchmarks above (reads with a median length of 5000) on a single A100 reduces
runtime by 2.6x from from 1h to 24min at the cost of increasing GPU memory
consumption. This would be different on different GPUs or with different read
lenghts. Some experimentation with data may be required to tune parameters.
Create a batch input file (e.g. guppy.sh). For example:
#!/bin/bash
set -e
module load guppy/6.1.2 || exit 1
guppy_basecaller --input_path $GUPPY_TEST_DATA/Zymo-GridION-EVEN-BB-SN --recursive \
--flowcell FLO-MIN106 --kit SQK-LSK109 \
-x cuda:all \
--records_per_fastq 0 \
--compress_fastq \
--save_path fastq
Submit this job using the Slurm sbatch command.
sbatch --partition=gpu --cpus-per-task=14 --mem=16g --gres=lscratch:200,gpu:p100:1 guppy.sh
Create a swarmfile (e.g. guppy.swarm). For example:
guppy_basecaller --input_path indir1 --flowcell FLO-MIN106 --kit SQK-LSK109 --save_path outdir1 ... guppy_basecaller --input_path indir2 --flowcell FLO-MIN106 --kit SQK-LSK109 --save_path outdir2 ... ...etc...
Submit this job using the swarm command.
swarm -f guppy.swarm --partition=gpu -g 16 -t 14 --gres=gpu:p100:1 --module guppy/3.2.2where
| -g # | Number of Gigabytes of memory required for each process (1 line in the swarm command file) |
| -t # | Number of threads/CPUs required for each process (1 line in the swarm command file). |
| --module guppy | Loads the guppy module for each subjob in the swarm |