Qualimap on Biowulf
Qualimap is a platform-independent application written in Java and R that provides both a Graphical User Inter- face (GUI) and a command-line interface to facilitate the quality control of alignment sequencing data.
References:
- Garcia-Alcalde et al.Qualimap: evaluating next-generation sequencing alignment data. 2012.Link
Documentation
Important Notes
- Module Name: qualimap (see the modules page for more information)
- Singlethreaded/X11 needed
- Example files in /fdb/app_testdata/bam/hg19/subsample.bam
Interactive job
Interactive jobs should be used for debugging, graphics, or applications that cannot be run as batch jobs.
You need to make an X-windows connection
to Helix to allow the Qualimap GUI to display on your local desktop. Then
type 'module load qualimap' to set up the environment, and then type 'qualimap'.
Allocate an interactive session and run the program. Sample session:
[user@biowulf]$ sinteractive --cpus-per-task=4 --mem=8g
salloc.exe: Pending job allocation 46116226
salloc.exe: job 46116226 queued and waiting for resources
salloc.exe: job 46116226 has been allocated resources
salloc.exe: Granted job allocation 46116226
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn3144 are ready for job
[user@cn3144 ~]$ module load qualimap
[+] Loading Perl 5.8.9 ...
[+] Loading gcc 4.4.7 ...
[+] Loading OpenMPI 1.8.1 for GCC 4.4.7 (ethernet) ...
[+] Loading tcl_tk 8.6.1
[+] Loading LAPACK 3.5.0-gcc-4.4.7 libraries...
[+] Loading R 3.2.0 on cn3144
[user@cn3144 ~]$ qualimap bamqc -nt $SLURM_CPUS_PER_TASK -bam test_DNase_seq.hg19.bam -outfile result.pdf
Java memory size is set to 1200M
Launching application...
QualiMap v.2.2
Built on 2016-01-29 12:10
Selected tool: bamqc
Available memory (Mb): 33
Max memory (Mb): 1118
Starting bam qc....
Loading sam header...
Loading locator...
Loading reference...
Number of windows: 400, effective number of windows: 423
Chunk of reads size: 1000
Number of threads: 4
Processed 50 out of 423 windows...
Processed 100 out of 423 windows...
Processed 150 out of 423 windows...
Processed 200 out of 423 windows...
Processed 250 out of 423 windows...
Processed 300 out of 423 windows...
Processed 350 out of 423 windows...
Processed 400 out of 423 windows...
Total processed windows:423
Number of reads: 4830586
Number of valid reads: 4830586
Number of correct strand reads:0
Inside of regions...
Num mapped reads: 4830586
Num mapped first of pair: 0
Num mapped second of pair: 0
Num singletons: 0
Time taken to analyze reads: 111
Computing descriptors...
numberOfMappedBases: 173901096
referenceSize: 3036320417
numberOfSequencedBases: 173854103
numberOfAs: 46212432
Computing per chromosome statistics...
Computing histograms...
Overall analysis time: 112
end of bam qc
Computing report...
Writing PDF report...
PDF file created successfully
Finished
[user@cn3144 ~]$ exit
salloc.exe: Relinquishing job allocation 46116226
[user@biowulf ~]$
Interactive GUI job on Biowulf
Qualimap uses some R packages. R cannot be run on the Biowulf login node. Therefore, to run the Qualimap GUI interactively, you need to allocate an interactive node. First make an Xwindows connection to Biowulf.
To increase the memory available to qualimap, use the '-java-mem-size' parameter. e.g.
qualimap --java-mem-size=8000M
qualimap bamqc -bam very_large_alignment.bam --java-mem-size=4G
Sample session:
[user@biowulf ~]$ sinteractive --mem=8g
salloc.exe: Granted job allocation 143331
[user@cn0124 ~]$ module load qualimap
[+] Loading Perl 5.8.9 ...
[+] Loading gcc 4.4.7 ...
[+] Loading OpenMPI 1.8.1 for GCC 4.4.7 (ethernet) ...
[+] Loading tcl_tk 8.6.1
[+] Loading LAPACK 3.5.0-gcc-4.4.7 libraries...
[+] Loading R 3.2.0 on cn0124
[user@cn0124 ~]$ qualimap --java-mem-size=8000M
Java memory size is set to 8000M
Launching application...
QualiMap v.2.1.1
Built on 2015-06-15 14:19
Qualimap home is /usr/local/apps/qualimap/qualimap_v2.1.1
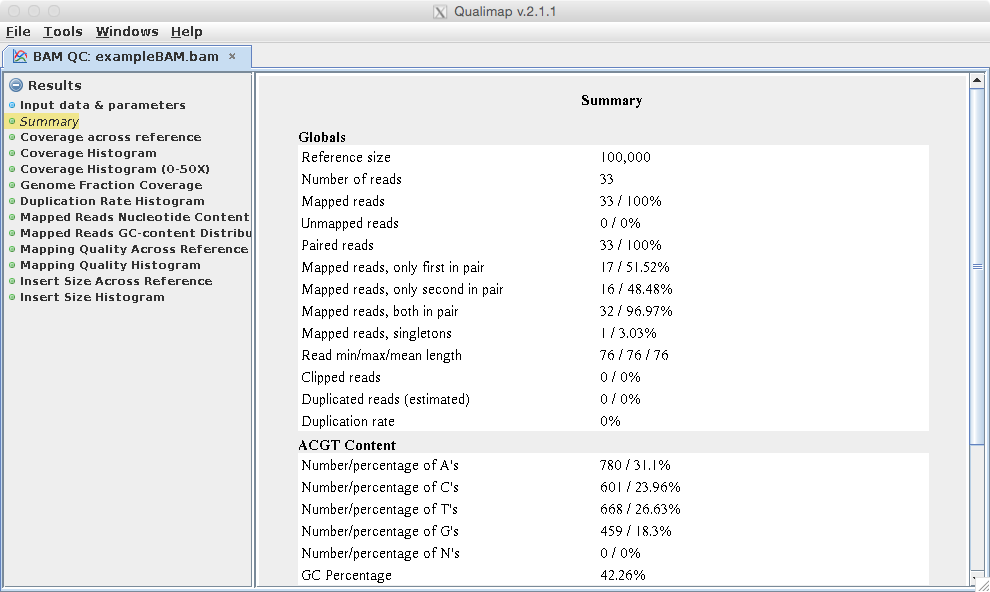
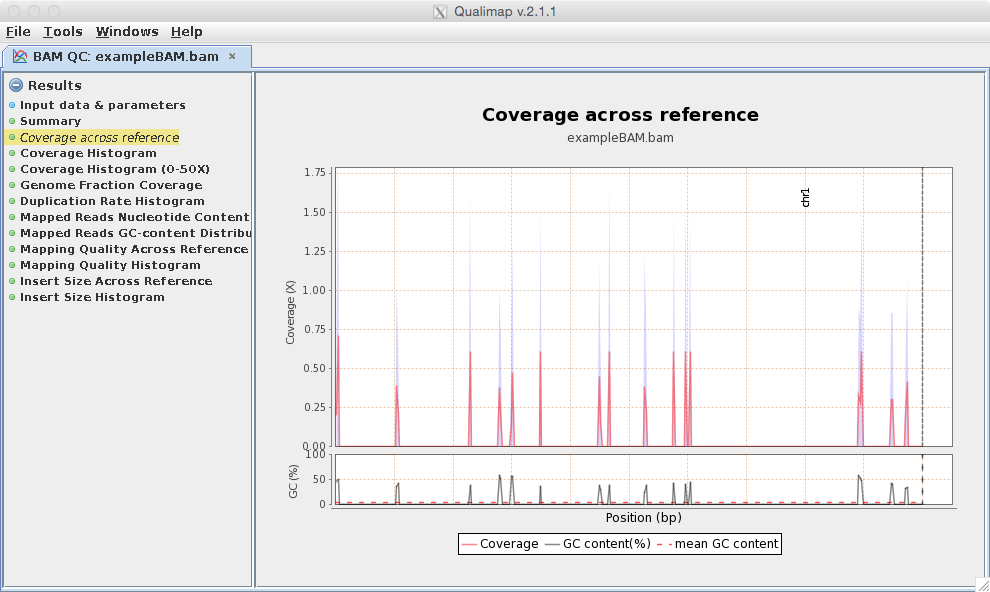 [user@cn0124 ~]$ exit
exit
salloc.exe: Relinquishing job allocation 143331
salloc.exe: Job allocation 143331 has been revoked.
[user@biowulf ~]$
[user@cn0124 ~]$ exit
exit
salloc.exe: Relinquishing job allocation 143331
salloc.exe: Job allocation 143331 has been revoked.
[user@biowulf ~]$
Batch job
Create a batch input file (e.g. qualimap.sh). For example:
#!/bin/bash
cd /data/$USER/mydir
module load qualimap
unset DISPLAY
qualimap bamqc -nt $SLURM_CPUS_PER_TASK -bam test_DNase_seq.hg19.bam -outfile result.pdf
Submit this job using the Slurm sbatch command.
sbatch --cpus-per-task=8 --mem=10g qualimap.sh
Important notes:
- By default, Qualimap's BAMQC module will auto-thread to utilize all CPUs on a node. Your job will then run multiple threads on each CPU, which is generally inefficient. Thus,
the -nt $SLURM_CPUS_PER_TASK flag has been set in the script to ensure that the number of threads matches the number of allocated CPUs. The other Qualimap modules,
e.g. RNA-seq, multi-bamqc etc do not multi-thread.
- If you're getting errors about 'Can't connect to X11 server', add
unset DISPLAY
to your batch script before the 'qualimap' command line to prevent the Java virtual machine from trying to use the X11 window system.
Swarm of Jobs
A
swarm of jobs is an easy way to submit a set of independent commands requiring identical resources.
Create a swarmfile (e.g. qualimap.swarm). For example:
unset DISPLAY; qualimap rnaseq -bam file1.bam -nt $SLURM_CPUS_PER_TASK -gtf Homo_sapiens.GRCh37.gtf -outdir rnaseq_qc_results
unset DISPLAY; qualimap rnaseq -bam file2.bam -nt $SLURM_CPUS_PER_TASK -gtf Homo_sapiens.GRCh37.gtf -outdir rnaseq_qc_results
Submit this job using the swarm command.
swarm -f qualimap.swarm -g 10 -t 8 --module qualimap
where
| -g # | Number of Gigabytes of memory required for each process (1 line in the swarm command file)
|
| -t # | Number of threads/CPUs required for each process (1 line in the swarm command file).
|
| --module qualimap | Loads the qualimap module for each subjob in the swarm
|