In the previous Data Storage hands-on section, you should have copied the class scripts to your /data area. If you skipped or missed that section, type
hpc-classes biowulfnow. This command will copy the scripts and input files used in this online class to your /data area, and will take about 5 minutes.
In the following session, you will submit a batch job for Bowtie2, a genome alignment program for short read sequences. If you're not a genomicist, don't worry -- this is just an example. The basic principles of job submission are not specific for Bowtie.
cd /data/$USER/hpc-classes/biowulf/bowtie # submit the job sbatch --cpus-per-task=16 bowtie.bat # check the status with 'jobload' jobload -u $USER # after the job finishes, use 'jobhist' to see how much memory it used # (replace 'jobnumber' in the command below with the job number of your bowtie job) jobhist jobnumber
Point your web browser to https://hpc.nih.gov/dashboard. You will need to log in with your NIH username and password. Click on the 'Job Info' tab.
You should see a list of your jobs from the last few days. Click on the job ID of the running or completed bowtie job, and you will see a plot showing the memory and CPU usage of the job. It should look something like this at the end:
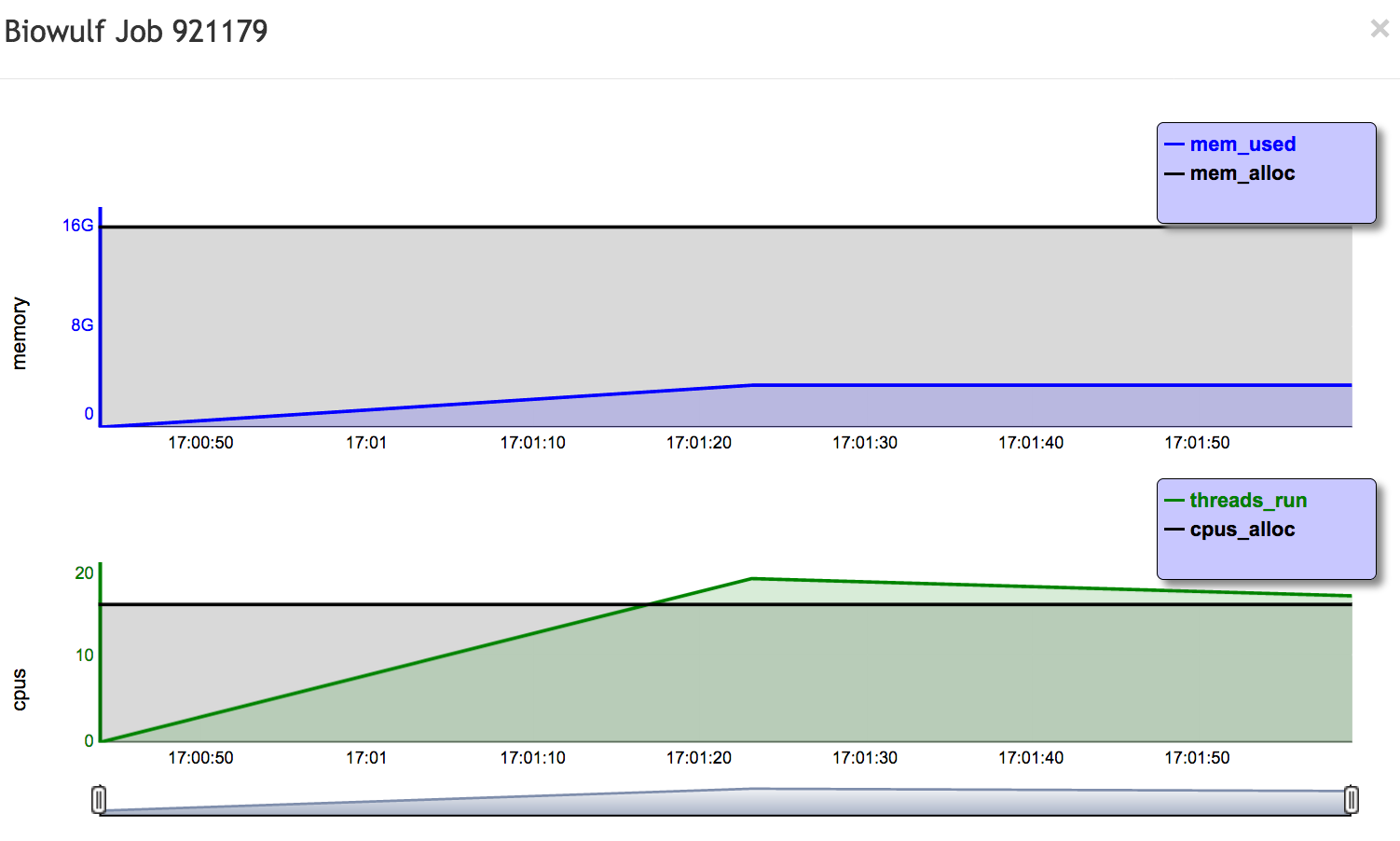
You can scroll down further and get additional information about the job.
Answer
sbatch --cpus-per-task=16 --mem=5g bowtie.bat
Answer
Answer
It might help to allocate more CPUs if the bowtie process scales well, and the job is not limited by I/O. Try it by submitting with
sbatch --cpus-per-task=32 --mem=5g bowtie.batAfter it completes, use 'jobhist jobnumber' to check the walltime. In my tests, I got the following walltimes:
So for this job, it is worth using 16 rather than 8 CPUs, given the speedup. But doubling the CPUs from 16 to 32 does not double the performance (i.e. halve the walltime), which suggests that I/O is a limiting factor. Thus, 16 CPUs is the sweet spot for this job.