Bowtie2 is a fast, multi-threaded, and memory efficient aligner for short read sequences. It uses an FM index to achieve a moderate memory footprint of 2 - 4 GB, depending on genome size and alignment parameters. Performance scales well with thread count.
Note that this page only describes bowtie2. Bowtie1 is described on a separate page. Unlike bowtie1, bowtie2 supports local alignments and gapped alignments, amongst other enhancements and new features. It is also more suited for longer reads and calculates a more informative MAPQ than bowtie1.
$BOWTIE_TEST_DATABowtie2 indices are available as part of the igenomes package under
/fdb/igenomes/[organism]/[source]/[build]/Sequence/Bowtie2Index/*
[organism] is the specific organism of interest
(Gallus_gallus, Rattus_norvegicus, etc.)[source] is the source for the sequence (NCBI,
Ensembl, UCSC)[build] is the specific genome build of interest
(hg19, build37.2, GRCh37)More information on the locally available igenomes builds/organisms is available from our scientific database index. For more information about igenomes in general, iGenomes readme.
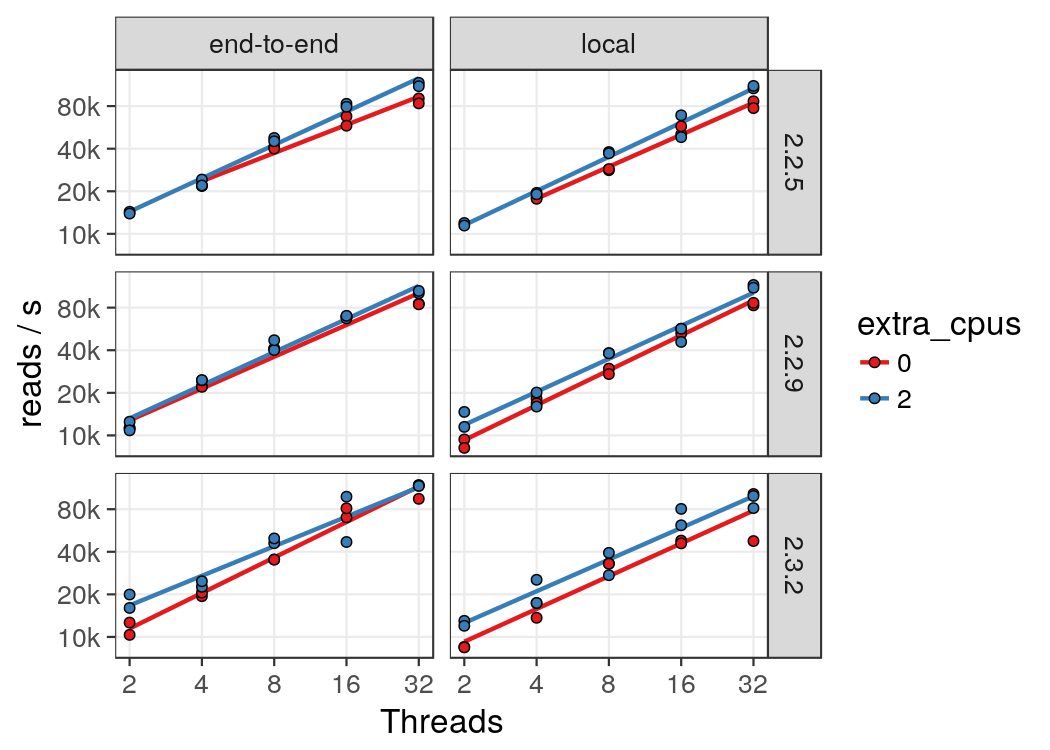
Bowtie2 is a multithreaded application. To determine how well its performace
scales with the number of threads, the same input file (the first 25M ChIP-Seq reads
from mouse heart tissue, ENCSR699XHY,
replicate 2) was aligned with different numbers of threads. Note that some versions of bowtie2
,in addition to the alignment threads, also starts some supporting processes such
as a process to expand compressed input. These processes can lead to reduced
efficiency due to frequent context switches (i.e. job overloading). To measure
the effect of this, the same experiment was carried out in two differnet conditions:
(1) In one case, bowtie2 was allowed to run as many threads as there were allocated
CPUs (--threads=$SLURM_CPUS_PER_TASK), which lead to a mild overload.
(2) In the other case, two extra CPUs were allocated (i.e.
threads=$(( SLURM_CPUS_PER_TASK - 2 ))) to account for the extra processes.
Local alignments were approximately 20% slower than end-to-end alignments. Both were done in sensitive mode for several versions of bowtie2.
From this we can see that allocating 2 CPUs more than there are bowtie2 threads can have a modest performance benefit. Also, bowtie2 scales linearly with the number of threads up to 32. However, the slope is less than 1 and therefore it is inefficient to run with more than 8-16 threads.
The following example aligns single ended data, creating bam output
directly and uncompressing gziped fastq on the fly. In this case, the
alignment mode is set to sensitive-local. ALignments are filtered to
remove any with MAPQ < 30.
Allocate an interactive session and run the program. Sample session:
[user@biowulf]$ sinteractive --gres=lscratch:10 --cpus-per-task=6
salloc.exe: Pending job allocation 46116226
salloc.exe: job 46116226 queued and waiting for resources
salloc.exe: job 46116226 has been allocated resources
salloc.exe: Granted job allocation 46116226
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn3144 are ready for job
[user@cn3144 ~]$
[user@cn3144 ~]$ module load bowtie/2
[user@cn3144 ~]$ module load samtools
[user@cn3144 ~]$ cd /lscratch/$SLURM_JOB_ID
[user@cn3144 ~]$ export BOWTIE2_INDEXES=/fdb/igenomes/Mus_musculus/UCSC/mm9/Sequence/Bowtie2Index/
[user@cn3144 ~]$ bowtie2 --phred64 -x genome --threads=$SLURM_CPUS_PER_TASK \
--no-unal --end-to-end --sensitive \
-U $BOWTIE_TEST_DATA/ENCFF001KPB.fastq.gz \
| samtools view -q30 -Sb - > ENCFF001KPB.bam
9157799 reads; of these:
9157799 (100.00%) were unpaired; of these:
772509 (8.44%) aligned 0 times
7080210 (77.31%) aligned exactly 1 time
1305080 (14.25%) aligned >1 times
[user@cn3144 ~]$ exit
salloc.exe: Relinquishing job allocation 46116226
[user@biowulf ~]$
Create a batch input file (e.g. bowtie2.sh), which uses the input file 'bowtie2.in'. For example:
#!/bin/bash
module load bowtie/2 || exit 1
module load samtools || exit 1
export BOWTIE2_INDEXES=/fdb/igenomes/Mus_musculus/UCSC/mm9/Sequence/Bowtie2Index/
bowtie2 --phred64 -x genome --threads=$(( SLURM_CPUS_PER_TASK - 4 )) \
--no-unal --end-to-end --sensitive \
-U $BOWTIE_TEST_DATA/ENCFF001KPB.fastq.gz \
| samtools view -q30 -u - \
| samtools sort -O BAM -@3 -T /lscratch/$SLURM_JOB_ID/ENCFF001KPB -m 2g -o ENCFF001KPB.bam
Submit this job using the Slurm sbatch command.
sbatch --cpus-per-task=10 --mem=14g --gres=lscratch:10 bowtie2.sh
Create a swarmfile (e.g. bowtie2.swarm). For example:
cd /data/$USER/test_data \
&& export BOWTIE2_INDEXES=/fdb/igenomes/Mus_musculus/UCSC/mm9/Sequence/Bowtie2Index \
&& bowtie2 --sensitive-local -p $(( SLURM_CPUS_PER_TASK - 2 )) --no-unal -x genome \
-U /usr/local/apps/bowtie/TEST_DATA/ENCFF001KPB.fastq.gz \
| samtools view -q30 -Sb - > ENCFF001KPB.bam
cd /data/$USER/test_data \
&& export BOWTIE2_INDEXES=/fdb/igenomes/Mus_musculus/UCSC/mm9/Sequence/Bowtie2Index \
&& bowtie2 --sensitive-local -p $(( SLURM_CPUS_PER_TASK - 2 )) --no-unal -x genome \
-U /usr/local/apps/bowtie/TEST_DATA/ENCFF322WUF.fastq.gz \
| samtools view -q30 -Sb - > ENCFF322WUF.bam
Submit this job using the swarm command.
swarm -f bowtie2.swarm -g 14 -t 10 --module bowtie/2,samtoolswhere
| -g # | Number of Gigabytes of memory required for each process (1 line in the swarm command file) |
| -t # | Number of threads/CPUs required for each process (1 line in the swarm command file). |
| --module bowtie2 | Loads the bowtie2 module for each subjob in the swarm |