Bowtie1 is a fast, multi-threaded, and memory efficient aligner for short read sequences. Bowtie uses a Burrows-Wheeler index to achieve a moderate memory footprint of 2 - 4 GB depending on genome size and alignment parameters. Performance generally scales well with thread count.
Note that this page only describes bowtie1. Bowtie2, which supports local alignment, gaps, and longer reads, is documented separately.
$BOWTIE_TEST_DATABowtie1 indices are available as part of the igenomes package under
/fdb/igenomes/[organism]/[source]/[build]/Sequence/BowtieIndex/*
[organism] is the specific organism of interest
(Gallus_gallus, Rattus_norvegicus, etc.)[source] is the source for the sequence (NCBI,
Ensembl, UCSC)[build] is the specific genome build of interest
(hg19, build37.2, GRCh37)More information on the locally available igenomes builds/organisms is available from our scientific database index. For more information about igenomes in general, iGenomes readme.
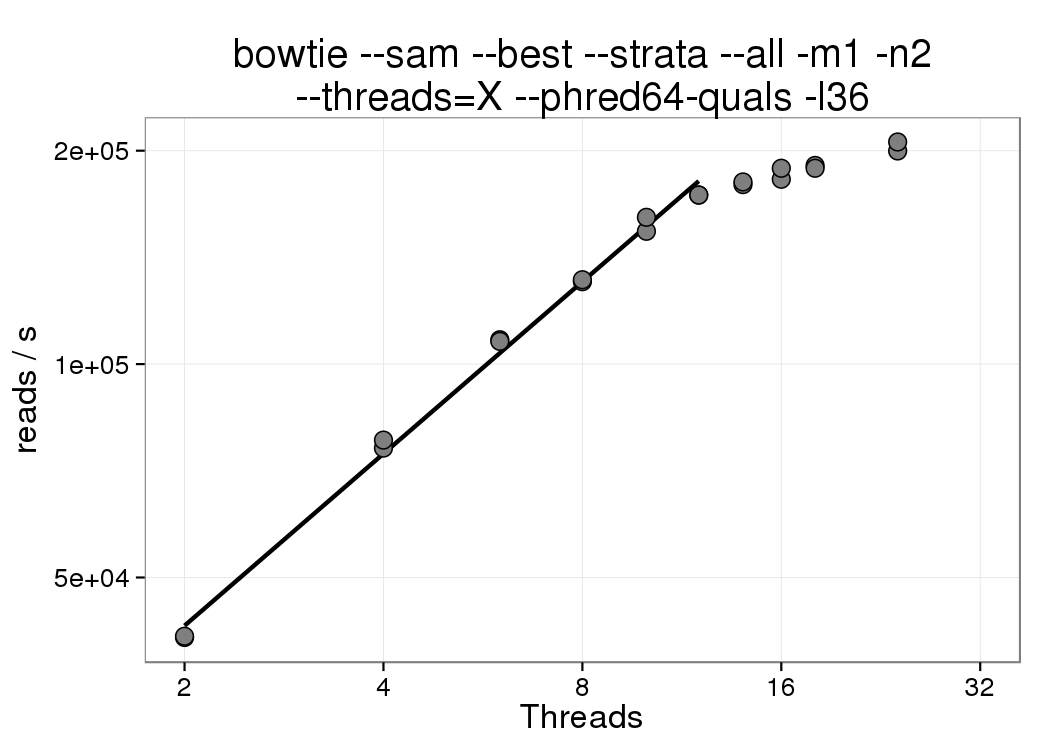
The amount of time to complete the alignment of approximately 21M ChIP-Seq reads (replicates 1 and 2 of ENCODE experiment ENCSR000CDI, 36nt, H3K27ac ChIP from mouse embryonic fibroblasts) was measured as a function of the number of bowtie threads:
Based on this experiment, increasing the number of threads to more than 12 shows diminishing returns. Therefore the most resource efficient usage of bowtie1 would employ at most 12 threads. If you are gunzipping input files and piping output through samtools, please allocate extra CPUs. Otherwise, the node will be slightly overloaded, which results in a considerable performance penalty due to contention between threads.
Allocate an interactive session and run the program. Sample session:
[user@biowulf]$ sinteractive --gres=lscratch:10 salloc.exe: Pending job allocation 46116226 salloc.exe: job 46116226 queued and waiting for resources salloc.exe: job 46116226 has been allocated resources salloc.exe: Granted job allocation 46116226 salloc.exe: Waiting for resource configuration salloc.exe: Nodes cn3144 are ready for job [user@cn3144 ~]$ module load bowtie/1 [user@cn3144 ~]$ module load samtools/1.6 [user@cn3144 ~]$ cd /lscratch/$SLURM_JOB_ID [user@cn3144 ~]$ export BOWTIE_INDEXES=/fdb/igenomes/Mus_musculus/UCSC/mm9/Sequence/BowtieIndex/ [user@cn3144 ~]$ ls $BOWTIE_INDEXES genome.1.ebwt genome.2.ebwt genome.3.ebwt genome.4.ebwt genome.fa genome.rev.1.ebwt genome.rev.2.ebwt [user@cn3144 ~]$ zcat $BOWTIE_TEST_DATA/ENCFF001KPB.fastq.gz \ | bowtie --phred64-quals --strata --best --all --chunkmbs 256 -m1 -n2 -p2 --sam -x genome - \ | samtools view -F4 -Sb - > ENCFF001KPB.bam # reads processed: 11623213 # reads with at least one reported alignment: 9467690 (81.46%) # reads that failed to align: 955092 (8.22%) # reads with alignments suppressed due to -m: 1200431 (10.33%) Reported 9467690 alignments [user@cn3144 ~]$ exit salloc.exe: Relinquishing job allocation 46116226 [user@biowulf ~]$
Create a batch input file (e.g. bowtie1.sh), which uses the input file 'bowtie1.in'. For example:
#!/bin/bash
module load bowtie/1 samtools || exit 1
wd=$PWD
export BOWTIE_INDEXES=/fdb/igenomes/Mus_musculus/UCSC/mm9/Sequence/BowtieIndex/
cd /lscratch/$SLURM_JOB_ID
zcat $BOWTIE_TEST_DATA/ENCFF001KPB.fastq.gz \
| bowtie --phred64-quals --strata --best --all --chunkmbs 256 -m1 -n2 -p${SLURM_CPUS_PER_TASK} --sam -x genome - \
| samtools view -F4 -Sb - > ENCFF001KPB.bam
mv ENCFF001KPB.bam $wd
Submit this job using the Slurm sbatch command.
sbatch --cpus-per-task=8 --mem=4g bowtie1.sh
Create a swarmfile (e.g. bowtie1.swarm). For example:
export BOWTIE_INDEXES=/fdb/igenomes/Mus_musculus/UCSC/mm9/Sequence/BowtieIndex/; \ zcat sample1.fastq.gz | bowtie --strata --best --all -m1 --sam -x genome - | samtools view -F4 -Sb - > sample1.bam export BOWTIE_INDEXES=/fdb/igenomes/Mus_musculus/UCSC/mm9/Sequence/BowtieIndex/; \ zcat sample2.fastq.gz | bowtie --strata --best --all -m1 --sam -x genome - | samtools view -F4 -Sb - > sample2.bam export BOWTIE_INDEXES=/fdb/igenomes/Mus_musculus/UCSC/mm9/Sequence/BowtieIndex/; \ zcat sample3.fastq.gz | bowtie --strata --best --all -m1 --sam -x genome - | samtools view -F4 -Sb - > sample3.bam
Submit this job using the swarm command.
swarm -f bowtie1.swarm -g 4 -t 8 --module bowtie/1,samtoolswhere
| -g # | Number of Gigabytes of memory required for each process (1 line in the swarm command file) |
| -t # | Number of threads/CPUs required for each process (1 line in the swarm command file). |
| --module bowtie1 | Loads the bowtie1 module for each subjob in the swarm |