
|
Quick Links
|
RELION (for REgularised LIkelihood OptimisatioN, pronounce rely-on) is a stand-alone computer program that employs an empirical Bayesian approach to refinement of (multiple) 3D reconstructions or 2D class averages in electron cryo-microscopy (cryo-EM).
RELION jobs MUST utilize local scratch in order to prevent filesystem performance degradation. If submitting through the GUI, PLEASE SET THE 'Copy particles to scratch directory' input to /lscratch/$SLURM_JOB_ID in the Compute tab:
Do not leave the Gres input empty. Minimally allocate 5 GB of lscratch in each job.

If running RELION jobs on multinode partition, it's mandatory to include --constraint(equivalent to -C) in the "Additional SBATCH Directives" input. See details on the multinode use

If submitting from a batch script, local scratch can be utilized by including the option --scratch_dir /lscratch/$SLURM_JOB_ID in the command line.
NOTE: Do not include --mem in batch allocations. The Slurm batch system cannot accept both --mem-per-cpu and --mem in submissions. RELION is best run with --mem-per-cpu only.
| PATH | RELION_QSUB_EXTRA1_DEFAULT | RELION_QSUB_EXTRA6_DEFAULT |
| RELION_CTFFIND_EXECUTABLE | RELION_QSUB_EXTRA2 | RELION_QSUB_EXTRA_COUNT |
| RELION_ERROR_LOCAL_MPI | RELION_QSUB_EXTRA2_DEFAULT | RELION_QSUB_NRMPI |
| RELION_GCTF_EXECUTABLE | RELION_QSUB_EXTRA3 | RELION_QSUB_NRTHREADS |
| RELION_HOME | RELION_QSUB_EXTRA3_DEFAULT | RELION_QSUB_TEMPLATE |
| RELION_MINIMUM_DEDICATED | RELION_QSUB_EXTRA4 | RELION_QUEUE_NAME |
| RELION_MOTIONCOR2_EXECUTABLE | RELION_QSUB_EXTRA4_DEFAULT | RELION_QUEUE_USE |
| RELION_MOTIONCORR_EXECUTABLE | RELION_QSUB_EXTRA5 | RELION_RESMAP_EXECUTABLE |
| RELION_MPI_MAX | RELION_QSUB_EXTRA5_DEFAULT | RELION_THREAD_MAX |
| RELION_QSUB_COMMAND | RELION_QSUB_EXTRA6 | RELION_VERSION |
| RELION_QSUB_EXTRA1 | RELION_STD_LAUNCHER | RELION_MPIRUN |
/usr/local/apps/RELION/mamba/envs/torch1.10/bin/python
Interactive use of RELION via the GUI requires an graphical X11 connection. HPC OnDemand works well, while XQuartz sometimes works for Mac users.
Start an interactive session on the Biowulf cluster. For example, this allocates 16 CPUs, 32GB of memory, 200GB of local scratch space, and 16 hours of time:
sinteractive --cpus-per-task=16 --mem-per-cpu=2g --gres=lscratch:200 --time=16:00:00
load the RELION module and start up the GUI:
[user@cn1234 ~]$ cd /path/to/your/RELION/project/ [user@cn1234 project]$ module load RELION [user@cn1234 project]$ relion
This should start the main GUI window:
Jobs that are suitable for running on the interactive host can be run directly from the GUI. For example, running CTF:
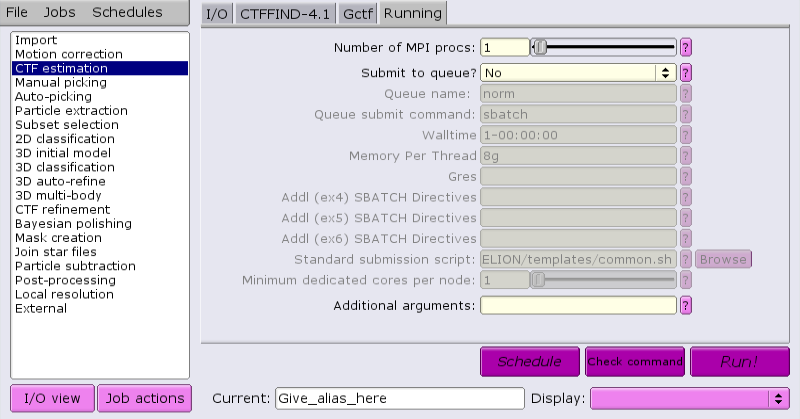
Once the job parameters are defined, just click 'Run now!'.
If the RELION process run on the local host of an interactive session is MPI-enabled, the number of MPI procs set in the GUI must match the number of tasks allocated for the job.
By default, an interactive session allocates a single task. This means that by default, only a single MPI proc can be run from the GUI. To start an interactive session with the capability of handling multiple MPI procs, add --ntasks and --nodes=1 to the sinteractive command, and adjust --cpus-per-task accordingly:
sinteractive --cpus-per-task=1 --nodes=1 --ntasks=16 --mem-per-cpu=2g --gres=lscratch:200 --time=16:00:00
Jobs that should be run on different host(s) can be launched from a generic interactive session and run on the batch system by choosing the appropriate parameters. The interactive session does not need elaborate resources, although it must be submitted from within a graphical X11 session on the login node.
sinteractive
Once the interactive session has started, the GUI can be launched from the project directory like so:
[user@cn1234 ~]$ cd /path/to/your/RELION/project/ [user@cn1234 project]$ module load RELION [user@cn1234 project]$ relion
Here is a job that will allocate 512 MPI tasks, each with 8 CPUs per task, for a total of 4096 CPUs. The CPUs will have the x2695 property, meaning they will be Intel E5-2695v3 processors. Each CPU will have access to 4 GB of RAM memory. Each node will have 400 GB of local scratch space available to the job, and the total time alloted for the job to complete is 2 days.
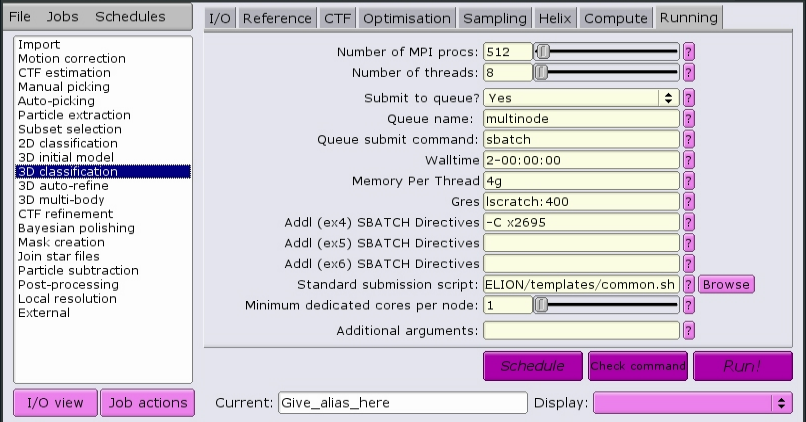
See Recommended parameters for a set of recommended parameters for each job type.
Alternatively, there are pre-made template scripts for most jobtypes available.
Choosing the appropriate parameters for GUI batch jobs on HPC Biowulf can be very complicated. Below is a relatively straightforward guide to those parameters based on job type.
In all cases below the amount of walltime allocated is an estimate. Your time may vary depending mainly on the number of particles and the number of MPI procs. More particles and fewer MPI procs means more time required.
Also, in all cases please set 'Queue submit command' to sbatch in the running tab. This is required for the batch job to be submitted to the biowulf cluster.
Listed below are recommended parameters for all nodetypes availble for RELION. It is usually best to allocate a single nodetype for a multinode job, as the processor speeds will be matched. The nodetypes are listed in order of increasing performance, but alas the faster the node, the more in demand it is and the longer you may need to wait.
Motion tab:
| Use RELION's own implementation? | no |
| MOTIONCOR2 executable: | /usr/local/apps/MotionCor2/1.3.0/MotionCor2 |
| Use GPU acceleration? | yes |
| Which GPUs to use: | -- leave blank -- |
Running tab:
| Number of MPI procs: | 4, 8, 12, 16, OR 20 (multiple of 4) |
| Number of threads: | 1 |
| Submit to queue? | yes |
| Queue name: | gpu |
| Walltime: | 8:00:00 |
| Memory Per Thread: | 20g |
| Gres: | gpu:p100:4 |
| SBATCH Directives: | --ntasks-per-node=4 |
Motion tab:
| Use RELION's own implementation? | no |
| MOTIONCOR2 executable: | /usr/local/apps/MotionCor2/1.3.0/MotionCor2 |
| Use GPU acceleration? | yes |
| Which GPUs to use: | -- leave blank -- |
Running tab:
| Number of MPI procs: | 4, 8, 12, 16, OR 20 (multiple of 4) |
| Number of threads: | 1 |
| Submit to queue? | yes |
| Queue name: | gpu |
| Walltime: | 8:00:00 |
| Memory Per Thread: | 20g |
| Gres: | gpu:v100:4 |
| SBATCH Directives: | --ntasks-per-node=4 |
Motion tab:
| Use RELION's own implementation? | no |
| MOTIONCOR2 executable: | /usr/local/apps/MotionCor2/1.3.0/MotionCor2 |
| Use GPU acceleration? | yes |
| Which GPUs to use: | -- leave blank -- |
Running tab:
| Number of MPI procs: | 4, 8, 12, 16, OR 20 (multiple of 4) |
| Number of threads: | 1 |
| Submit to queue? | yes |
| Queue name: | gpu |
| Walltime: | 8:00:00 |
| Memory Per Thread: | 20g |
| Gres: | gpu:v100x:4 |
| SBATCH Directives: | --ntasks-per-node=4 |
Motion tab:
| Use RELION's own implementation? | no |
| MOTIONCOR2 executable: | /usr/local/apps/MotionCor2/1.3.0/MotionCor2 |
| Use GPU acceleration? | yes |
| Which GPUs to use: | -- leave blank -- |
Running tab:
| Number of MPI procs: | 4, 8, 12, 16, OR 20 (multiple of 4) |
| Number of threads: | 1 |
| Submit to queue? | yes |
| Queue name: | gpu |
| Walltime: | 8:00:00 |
| Memory Per Thread: | 15g |
| Gres: | gpu:a100:4 |
| SBATCH Directives: | --ntasks-per-node=4 |
NOTE 1: Consider using RELION's own motion correction implementation (below). On the average, it takes ~10x longer to allocate 8 GPUs than it does 512 CPUs.
NOTE 2: There are other versions of MotionCor2. To use these, load the different MotionCor2 module after loading the RELION module, but before running the relion command.
NOTE 3: The amount of memory shown above is minimum. The amount of memory required may be increased, but it depends on how many MPI procs are allocated and the total amount of memory available to the node. For example, p100 nodes have 117g total, and so with 4 MPI procs, the maximum memory would be (117/4), or ~29g. For 8 MPI procs, it would be (117/8), or ~14g.
Motion tab:
| Use RELION's own implementation? | yes |
| Use GPU acceleration? | no |
Running tab:
| Number of MPI procs: | 128-2048 |
| Number of threads: | 8 |
| Submit to queue? | yes |
| Queue name: | multinode |
| Walltime: | 8:00:00 |
| Memory Per Thread: | 16g |
| Gres: | lscratch:1 |
| SBATCH Directives: | --constraint=x2695 |
Motion tab:
| Use RELION's own implementation? | yes |
| Use GPU acceleration? | no |
Running tab:
| Number of MPI procs: | 128-2048 |
| Number of threads: | 8 |
| Submit to queue? | yes |
| Queue name: | multinode |
| Walltime: | 8:00:00 |
| Memory Per Thread: | 16g |
| Gres: | lscratch:1 |
| SBATCH Directives: | --constraint=x2680 |
NOTE 1: RELION's own implementation can require a large amount of memory per thread. If the job fails with memory errors, likely you will need to increase the amount beyond what is given above. However, the number of threads may need to be decreased. Memory usage can be monitored in the dashboard.
NOTE 2: Likely the job will complete in a few hours. If you want it to complete sooner, you can increase the number of MPI procs. However, the more you request, the longer the job will sit waiting for resources to become available.
CTFFIND-4.1 tab:
| Use CTFFIND-4.1? | yes |
| CTFFIND-4.1 executable: | /usr/local/apps/ctffind/4.1.14/ctffind |
Gctf tab:
| Use Gctf instead? | no |
Running tab:
| Number of MPI procs: | 128 |
| Submit to queue? | yes |
| Queue name: | multinode |
| Walltime: | 2:00:00 |
| Memory Per Thread: | 1g |
| Gres: | lscratch:1 |
CTFFIND-4.1 tab:
| Use CTFFIND-4.1? | no |
Gctf tab:
| Use Gctf instead? | no |
| Gctf executable: | /usr/local/apps/Gctf/1.06/bin/Gctf |
| Which GPUs to use: | -- leave blank -- |
Running tab:
| Number of MPI procs: | 4, 8, 12, 16, OR 20 (multiple of 4) |
| Number of threads: | 1 |
| Submit to queue? | yes |
| Queue name: | gpu |
| Walltime: | 8:00:00 |
| Memory Per Thread: | 20g |
| Gres: | gpu:p100:4 |
| SBATCH Directives: | --ntasks-per-node=4 |
CTFFIND-4.1 tab:
| Use CTFFIND-4.1? | no |
Gctf tab:
| Use Gctf instead? | no |
| Gctf executable: | /usr/local/apps/Gctf/1.06/bin/Gctf |
| Which GPUs to use: | -- leave blank -- |
Running tab:
| Number of MPI procs: | 4, 8, 12, 16, OR 20 (multiple of 4) |
| Number of threads: | 1 |
| Submit to queue? | yes |
| Queue name: | gpu |
| Walltime: | 8:00:00 |
| Memory Per Thread: | 20g |
| Gres: | gpu:v100:4 |
| SBATCH Directives: | --ntasks-per-node=4 |
CTFFIND-4.1 tab:
| Use CTFFIND-4.1? | no |
Gctf tab:
| Use Gctf instead? | no |
| Gctf executable: | /usr/local/apps/Gctf/1.06/bin/Gctf |
| Which GPUs to use: | -- leave blank -- |
Running tab:
| Number of MPI procs: | 4, 8, 12, 16, OR 20 (multiple of 4) |
| Number of threads: | 1 |
| Submit to queue? | yes |
| Queue name: | gpu |
| Walltime: | 8:00:00 |
| Memory Per Thread: | 20g |
| Gres: | gpu:v100x:4 |
| SBATCH Directives: | --ntasks-per-node=4 |
CTFFIND-4.1 tab:
| Use CTFFIND-4.1? | no |
Gctf tab:
| Use Gctf instead? | no |
| Gctf executable: | /usr/local/apps/Gctf/1.06/bin/Gctf |
| Which GPUs to use: | -- leave blank -- |
Running tab:
| Number of MPI procs: | 16, 32, 48, 64, OR 72 (multiple of 16) |
| Number of threads: | 1 |
| Submit to queue? | yes |
| Queue name: | gpu |
| Walltime: | 8:00:00 |
| Memory Per Thread: | 15g |
| Gres: | gpu:a100:4 |
| SBATCH Directives: | --ntasks-per-node=16 |
NOTE 1: Consider using RELION's own motion correction implementation (below). On the average, it takes ~10x longer to allocate 8 GPUs than it does 512 CPUs.
Compute tab:
| Copy particles to scratch directory: | /lscratch/$SLURM_JOB_ID |
| Use GPU acceleration? | yes |
| Which GPUs to use: | -- leave blank -- |
Running tab:
| Number of MPI procs: | 20 |
| Number of threads: | 2 |
| Submit to queue? | yes |
| Queue name: | gpu |
| Walltime: | 8:00:00 |
| Memory Per Thread: | 0 |
| Gres: | lscratch:650,gpu:p100:4 |
| SBATCH Directives: | --nodes=4 --ntasks-per-node=5 --exclusive |
Compute tab:
| Copy particles to scratch directory: | /lscratch/$SLURM_JOB_ID |
| Use GPU acceleration? | yes |
| Which GPUs to use: | -- leave blank -- |
Running tab:
| Number of MPI procs: | 20 |
| Number of threads: | 4 |
| Submit to queue? | yes |
| Queue name: | gpu |
| Walltime: | 8:00:00 |
| Memory Per Thread: | 0 |
| Gres: | lscratch:800,gpu:v100:4 |
| SBATCH Directives: | --nodes=4 --ntasks-per-node=5 --exclusive |
Compute tab:
| Copy particles to scratch directory: | /lscratch/$SLURM_JOB_ID |
| Use GPU acceleration? | yes |
| Which GPUs to use: | -- leave blank -- |
Running tab:
| Number of MPI procs: | 20 |
| Number of threads: | 4 |
| Submit to queue? | yes |
| Queue name: | gpu |
| Walltime: | 8:00:00 |
| Memory Per Thread: | 0 |
| Gres: | lscratch:1600,gpu:v100x:4 |
| SBATCH Directives: | --nodes=4 --ntasks-per-node=5 --exclusive |
Compute tab:
| Copy particles to scratch directory: | /lscratch/$SLURM_JOB_ID |
| Use GPU acceleration? | yes |
| Which GPUs to use: | -- leave blank -- |
Running tab:
| Number of MPI procs: | 20 |
| Number of threads: | 4 |
| Submit to queue? | yes |
| Queue name: | gpu |
| Walltime: | 8:00:00 |
| Memory Per Thread: | 0 |
| Gres: | lscratch:3200,gpu:a100:4 |
| SBATCH Directives: | --nodes=4 --ntasks-per-node=5 --exclusive |
NOTE 1: The number of MPI procs = (ntasks-per-node X nodes). See here for more details.
NOTE 2: It is critical that enough local scratch is allocated to accomodate the particle data. 400 GB is the minimum, it can be larger.
NOTE 3: Increasing the number of threads might lower the amount of time required, but at the risk of overloading the GPUs and causing the job the stall. See here for more information.
NOTE 4: If the value of --ntasks-per-node is odd, greater than 2, and GPUs are allocated across multiple nodes, then the template script will manually override the distribution of MPI tasks across the allocated cpus (see below).
NOTE 5: Setting Memory Per Thread to 0 (zero) and including --exclusive will cause slurm to allocate all memory on the node to the job.
Compute tab:
| Copy particles to scratch directory: | /lscratch/$SLURM_JOB_ID |
| Use GPU acceleration? | no |
Running tab:
| Number of MPI procs: | 128-2048 |
| Number of threads: | 8 |
| Submit to queue? | yes |
| Queue name: | multinode |
| Walltime: | 8:00:00 |
| Memory Per Thread: | 4200m |
| Gres: | lscratch:400 |
| SBATCH Directives: | --constraint=x2695 |
Compute tab:
| Copy particles to scratch directory: | /lscratch/$SLURM_JOB_ID |
| Use GPU acceleration? | no |
Running tab:
| Number of MPI procs: | 128-2048 |
| Number of threads: | 8 |
| Submit to queue? | yes |
| Queue name: | multinode |
| Walltime: | 8:00:00 |
| Memory Per Thread: | 4200m |
| Gres: | lscratch:800 |
| SBATCH Directives: | --constraint=x2680 |
NOTE 1: The amount of memory per thread may need to be larger, depending on the size of particle. Memory usage can be monitored in the dashboard.
NOTE 2: The larger the number of MPI procs, the sooner the job will complete. However, the more you request, the longer the job will sit waiting for resources to become available.
NOTE 3: In tests, increasing the number of threads per MPI proc above 8 has not shown to significantly decrease running time.
Compute tab:
| Copy particles to scratch directory: | /lscratch/$SLURM_JOB_ID |
| Use GPU acceleration? | no |
Running tab:
| Number of MPI procs: | 65 |
| Number of threads: | 16 |
| Submit to queue? | yes |
| Queue name: | multinode |
| Walltime: | 2-00:00:00 |
| Memory Per Thread: | 4g |
| Gres: | lscratch:400 |
NOTE 1:The amount of time required and memory needed is greatly reduced by increasing the number of threads per MPI proc. 16 is likely the highest possible number before complications occur.
NOTE 2:The number of MPI procs can be increased, but it must be an odd number.
Version 4 of RELION introduces VDAM (Variable-metric gradient Descent algorithm with Adaptive Moments estimation) optimization for use within 2D classification and initial model generation. This methodology requires a different set of parameters than with the old SAGD optimization. Principally VDAM does not rely upon MPI for compute acceleration, but instead makes use of single node multithreading.
Because VDAM requires GPUs, there are alternative sets of recommended parameters, depending on the node type:
Compute tab:
| Copy particles to scratch directory: | /lscratch/$SLURM_JOB_ID |
| Use GPU acceleration? | yes |
| Which GPUs to use: | -- leave blank -- |
Running tab:
| Number of MPI procs: | 1 |
| Number of threads: | 8 |
| Submit to queue? | yes |
| Queue name: | gpu |
| Walltime: | 2-00:00:00 |
| Memory Per Thread: | 0 |
| Gres: | lscratch:650,gpu:p100:4 |
| SBATCH Directives: | --exclusive |
Compute tab:
| Copy particles to scratch directory: | /lscratch/$SLURM_JOB_ID |
| Use GPU acceleration? | yes |
| Which GPUs to use: | -- leave blank -- |
Running tab:
| Number of MPI procs: | 1 |
| Number of threads: | 16 |
| Submit to queue? | yes |
| Queue name: | gpu |
| Walltime: | 2-00:00:00 |
| Memory Per Thread: | 0 |
| Gres: | lscratch:800,gpu:v100:4 |
| SBATCH Directives: | --exclusive |
Compute tab:
| Copy particles to scratch directory: | /lscratch/$SLURM_JOB_ID |
| Use GPU acceleration? | yes |
| Which GPUs to use: | -- leave blank -- |
Running tab:
| Number of MPI procs: | 1 |
| Number of threads: | 16 |
| Submit to queue? | yes |
| Queue name: | gpu |
| Walltime: | 2-00:00:00 |
| Memory Per Thread: | 0 |
| Gres: | lscratch:1600,gpu:v100x:4 |
| SBATCH Directives: | --exclusive |
Compute tab:
| Copy particles to scratch directory: | /lscratch/$SLURM_JOB_ID |
| Use GPU acceleration? | yes |
| Which GPUs to use: | -- leave blank -- |
Running tab:
| Number of MPI procs: | 1 |
| Number of threads: | 16 |
| Submit to queue? | yes |
| Queue name: | gpu |
| Walltime: | 2-00:00:00 |
| Memory Per Thread: | 0 |
| Gres: | lscratch:3200,gpu:a100:4 |
| SBATCH Directives: | --exclusive |
For subtomogram analysis, it is required to construct the individual pseudo-subtomogram particles, equivalent to the particle extraction process in the SPA workflow. This process can be acclerated by distributing the work across nodes using MPI. This does not require GPUs.
Running tab:
| Number of MPI procs: | 2-32 |
| Number of threads: | 16 |
| Submit to queue? | yes |
| Queue name: | multinode |
| Walltime: | 1:00:00 |
| Memory Per Thread: | 4g |
| Gres: | lscratch:400 |
NOTE 1:The number of MPI procs should not exceed the number of tomograms.
This step generates a de novo model without any prior knowledge. It does not use MPI, but can utilize GPUs with multiple threads on a single node.
Compute tab:
| Copy particles to scratch directory: | /lscratch/$SLURM_JOB_ID |
| Use GPU acceleration? | yes |
| Which GPUs to use: | -- leave blank -- |
Running tab:
| Number of MPI procs: | 1 |
| Number of threads: | 16 |
| Submit to queue? | yes |
| Queue name: | gpu |
| Walltime: | 1-00:00:00 |
| Memory Per Thread: | 0 |
| Gres: | lscratch:650,gpu:p100:4 |
| SBATCH Directives: | --exclusive |
Compute tab:
| Copy particles to scratch directory: | /lscratch/$SLURM_JOB_ID |
| Use GPU acceleration? | yes |
| Which GPUs to use: | -- leave blank -- |
Running tab:
| Number of MPI procs: | 1 |
| Number of threads: | 32 |
| Submit to queue? | yes |
| Queue name: | gpu |
| Walltime: | 1-00:00:00 |
| Memory Per Thread: | 0 |
| Gres: | lscratch:800,gpu:v100:4 |
| SBATCH Directives: | --exclusive |
Compute tab:
| Copy particles to scratch directory: | /lscratch/$SLURM_JOB_ID |
| Use GPU acceleration? | yes |
| Which GPUs to use: | -- leave blank -- |
Running tab:
| Number of MPI procs: | 1 |
| Number of threads: | 32 |
| Submit to queue? | yes |
| Queue name: | gpu |
| Walltime: | 1-00:00:00 |
| Memory Per Thread: | 0 |
| Gres: | lscratch:1600,gpu:v100x:4 |
| SBATCH Directives: | --exclusive |
Compute tab:
| Copy particles to scratch directory: | /lscratch/$SLURM_JOB_ID |
| Use GPU acceleration? | yes |
| Which GPUs to use: | -- leave blank -- |
Running tab:
| Number of MPI procs: | 1 |
| Number of threads: | 64 |
| Submit to queue? | yes |
| Queue name: | gpu |
| Walltime: | 1-00:00:00 |
| Memory Per Thread: | 0 |
| Gres: | lscratch:3200,gpu:a100:4 |
| SBATCH Directives: | --exclusive |
There is one pre-made sbatch template file, /usr/local/apps/RELION/templates/common.sh, as set by the environment variable $RELION_QSUB_TEMPLATE.
#!/bin/bash
#SBATCH --ntasks=XXXmpinodesXXX
#SBATCH --partition=XXXqueueXXX
#SBATCH --cpus-per-task=XXXthreadsXXX
#SBATCH --error=XXXerrfileXXX
#SBATCH --output=XXXoutfileXXX
#SBATCH --open-mode=append
#SBATCH --time=XXXextra1XXX
#SBATCH --mem-per-cpu=XXXextra2XXX
#SBATCH --gres=XXXextra3XXX
#SBATCH XXXextra4XXX
#SBATCH XXXextra5XXX
#SBATCH XXXextra6XXX
source add_extra_MPI_task.sh
env | sort
${RELION_STD_LAUNCHER} --mem-per-cpu=XXXextra2XXX XXXcommandXXX
By including SBATCH directives in the GUI, all combinations of resources are possible with the single script.
Additionally, there are template scripts for most job types available. These are located within subdirectories of the main template directory:
$ ls /usr/local/apps/RELION/templates/ AutoPick_gpu Class3D_cpu CtfFind InitialModel MotCorRel Refine3D_cpu x2680_exclusive.sh Class2D_cpu Class3D_gpu CtfRefine LocalRes MotionCor2 Refine3D_gpu x2695_exclusive.sh Class2D_gpu common.sh GCTF lscratch_mon.sh Polish
Each job type subdirectory has a set of template scripts that correspond to a particular scale of the job:
$ ls /usr/local/apps/RELION/templates/Class2D_cpu/ 01_tiny.sh 02_small.sh 03_medium.sh 04_large.sh 05_xlarge.sh
If one of these pre-made template scripts are used, the only input box in the Running tab of the RELION GUI that needs to be set is "Queue submit command".
There are also three template scripts for running CPU-only jobs exclusively (x2680_exclusive.sh, x2695_exclusive.sh). These scripts set memory and lscratch, and allow MPI and thread changes.
User-created template scripts can be substituted into the 'Standard submission script' box under the Running tab.

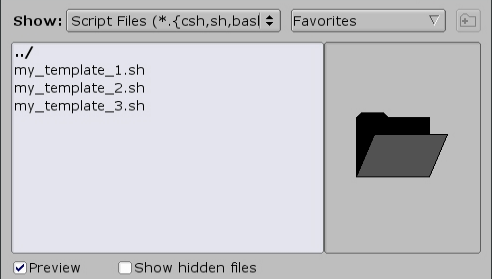
Alternatively, other templates can be browsed by clicking the 'Browse' button:
If the value of --ntasks-per-node is odd and greater than 2, and GPUs are allocated across multiple nodes, then the add_extra_MPI_task.sh script will generate a file ($SLURM_HOSTFILE) and change the distribution mode to arbitrary, manually overriding the distribution of MPI tasks across the allocated cpus:
#!/bin/bash
# Don't bother unless nodes have been allocated
if [[ -z $SLURM_JOB_NODELIST ]]; then
[[ -n $SLURM_HOSTFILE ]] && unset SLURM_HOSTFILE
return
fi
# Don't bother unless nodes have GPUs
if [[ -z $SLURM_JOB_GPUS ]]; then
[[ -n $SLURM_HOSTFILE ]] && unset SLURM_HOSTFILE
return
fi
# Don't bother unless multiple tasks have been allocated, and the number is odd
if [[ -z $SLURM_NTASKS_PER_NODE ]]; then
[[ -n $SLURM_HOSTFILE ]] && unset SLURM_HOSTFILE
return
elif [[ ${SLURM_NTASKS_PER_NODE} -lt 2 ]]; then
[[ -n $SLURM_HOSTFILE ]] && unset SLURM_HOSTFILE
return
elif [[ $((SLURM_NTASKS_PER_NODE%2)) == 0 ]]; then
[[ -n $SLURM_HOSTFILE ]] && unset SLURM_HOSTFILE
return
fi
# Don't bother unless there is more than one node
array=( $( scontrol show hostname $SLURM_JOB_NODELIST) )
file=$(mktemp --suffix .SLURM_JOB_NODELIST)
if [[ ${#array[@]} -eq 1 ]]; then
for ((j=0;j<$((SLURM_NTASKS_PER_NODE));j++)); do
echo ${array[0]} >> $file
done
else
echo ${array[0]} > $file
for ((i=0;i<${SLURM_JOB_NUM_NODES};i++)); do
for ((j=0;j<$((SLURM_NTASKS_PER_NODE-1));j++)); do
echo ${array[${i}]} >> $file
done
done
fi
# All conditions met, set hostfile and distribution, unset ntasks per node
export SLURM_HOSTFILE=$file
export SLURM_DISTRIBUTION=arbitrary
unset SLURM_NTASKS_PER_NODE
Allocate an interactive session and run the program.
Sample session (user input in bold):
[user@biowulf]$ sinteractive --gres=gpu:p100:4 --ntasks=4 --nodes=1 --ntasks-per-node=4 --mem-per-cpu=4g --cpus-per-task=2
salloc.exe: Pending job allocation 123457890
salloc.exe: job 1234567890 queued and waiting for resources
salloc.exe: job 1234567890 has been allocated resources
salloc.exe: Granted job allocation 1234567890
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn1234 are ready for job
[user@cn1234 ~]$ module load RELION
[user@cn1234 ~]$ ln -s /fdb/app_testdata/cryoEM/RELION/tutorials/relion31_tutorial_precalculated_results/Movies .
[user@cn1234 ~]$ mkdir import
[user@cn1234 ~]$ relion_import --do_movies --optics_group_name "opticsGroup1" --angpix 0.885 --kV 200 \
--Cs 1.4 --Q0 0.1 --beamtilt_x 0 --beamtilt_y 0 --i "Movies/*.tif" --odir Import --ofile movies.star
[user@cn1234 ~]$ mkdir output
[user@cn1234 ~]$ $RELION_MPIRUN relion_run_motioncorr_mpi --i Import/job001/movies.star --first_frame_sum 1 \
--last_frame_sum 0 --use_motioncor2 --motioncor2_exe ${RELION_MOTIONCOR2_EXECUTABLE} --bin_factor 1 \
--bfactor 150 --dose_per_frame 1.277 --preexposure 0 --patch_x 5 --patch_y 5 --gainref Movies/gain.mrc \
--gain_rot 0 --gain_flip 0 --dose_weighting --gpu '' --o output/
...
...
[user@cn1234 ~]$ exit
salloc.exe: Relinquishing job allocation 1234567890
[user@biowulf ~]$
Please note:
Allocating more than one node for an interactive, command-line driven RELION process will NOT run. Multi-node jobs must be submitted to the batch system.
Typically RELION batch jobs are submitted from the GUI. However, for those who insist on doing it themselves, here is an example of a batch input file (e.g. RELION.sh).
#!/bin/bash
#SBATCH --ntasks=20
#SBATCH --nodes=4
#SBATCH --ntasks-per-node=5
#SBATCH --cpus-per-task=2
#SBATCH --mem-per-cpu=6g
#SBATCH --partition=gpu
#SBATCH --gres=gpu:p100:4,lscratch:200
#SBATCH --error=run.err
#SBATCH --output=run.out
#SBATCH --time=1-00:00:00
module load RELION
source add_extra_MPI_task.sh
mkdir output
ln -s /fdb/app_testdata/cryoEM/plasmodium_ribosome/Particles .
ln -s /fdb/app_testdata/cryoEM/plasmodium_ribosome/emd_2660.map .
${RELION_STD_LAUNCHER} relion_refine_mpi \
--i Particles/shiny_2sets.star \
--o output/run \
--ref emd_2660.map:mrc \
--ini_high 60 \
--pool 100 \
--pad 2 \
--ctf \
--ctf_corrected_ref \
--iter 25 \
--tau2_fudge 4 \
--particle_diameter 360 \
--K 4 \
--flatten_solvent \
--zero_mask \
--oversampling 1 \
--healpix_order 2 \
--offset_range 5 \
--offset_step 2 \
--sym C1 \
--norm \
--scale \
--j 1
--gpu "" \
--dont_combine_weights_via_disc \
--scratch_dir /lscratch/${SLURM_JOB_ID}
Submit this job using the Slurm sbatch command.
sbatch RELION.sh
In order to understand how RELION accelerates its computation, we must understand two different concepts.
Multi-threading is when an executing process spawns multiple threads, or subprocesses, which share the same common memory space, but occupy independent CPUs.
Distributed tasks is the coordination of multiple independent processes by a single "leader" process via a communication protocol. MPI, or message passing interface, is the protocol by which these independent tasks are coordinated within RELION.
An MPI task can multi-thread, since each task is itself an independent process.
While all RELION job types can run with a single task in single-threaded mode, some can distribute their tasks via MPI. And a subset of those job types can further accelerate their computation by running those MPI tasks in multi-threaded mode.
For example, the Import job type can only run single task, single-threaded:
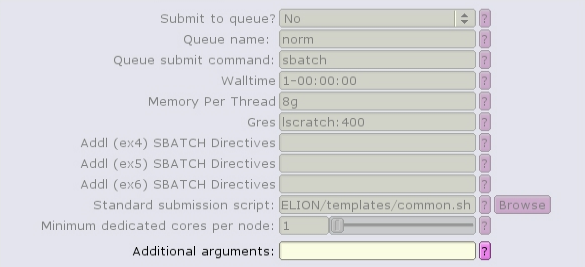
The CTF job type can run with multiple, distributed tasks, but each single-threaded:
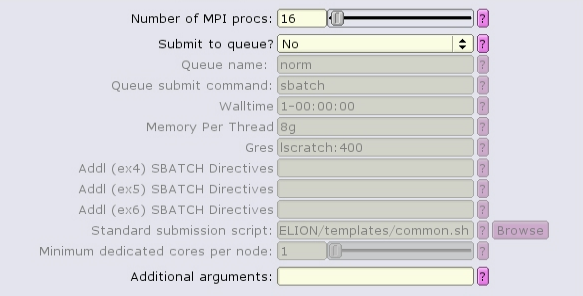
The MotionCor2 job type can run with multiple distributed tasks, each of which can run multi-threaded:
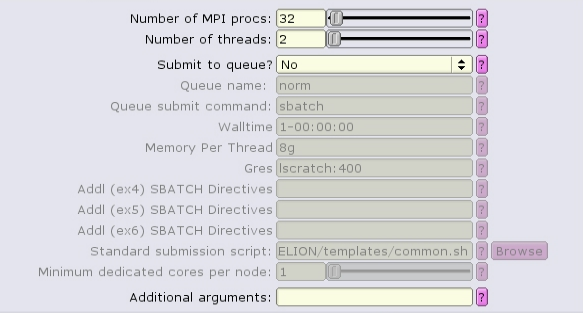
There are separate, distinct executables for running single-task and multi-task mode! If the "Number of MPI procs" value is left as one, then the single-task executable will be used:
relion_run_motioncorr --i import/movies.star --o output/ ...
If the value of "Number of MPI procs" is set to a value greater than one, then the MPI-enabled, distributed task executable will be used:
relion_run_motioncorr_mpi --i import/movies.star --o output/ ...
MPI-enabled executables must be launched properly to ensure proper distribution! When running in batch on the HPC cluster, the MPI-enabled executable should be launched with srun. This allows the MPI-enabled executable to discover what CPUs and nodes are available for tasks based on the Slurm environment:
srun relion_run_motioncorr_mpi --i import/movies.star --o output/ ...
RELION jobs using MPI-enabled executables can distribute their MPI tasks in one of three modes:
Certain job types benefit from these distributions. Classification jobs run on GPU nodes should use homogenous+1 distribution, while motion correction using MotionCor2 or GCTF should use homogenous distribution. Jobs run on CPU-only nodes can use heterogeneous distribution.
The distribution mode is dictated by additional SBATCH directives set in the 'Running' tab.
Because the number of nodes and the distribution of MPI tasks on those nodes is not known prior to submission, it is best to set the amount of memory allocated as Memory Per Thread, or --mem-per-cpu in the batch script.
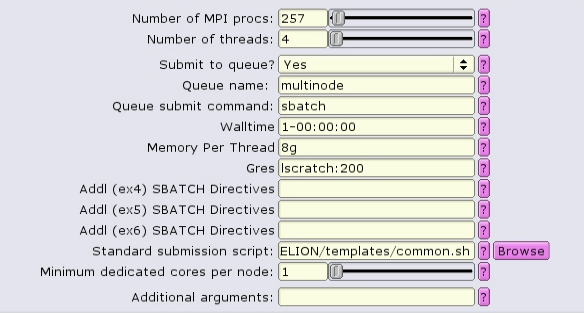
#!/bin/bash #SBATCH --ntasks=257 #SBATCH --partition=multinode #SBATCH --cpus-per-task=4 #SBATCH --error=run.err #SBATCH --output=run.out #SBATCH --time=1-00:00:00 #SBATCH --mem-per-cpu=8g #SBATCH --gres=lscratch:200 ... RELION command here ...
Visually, this distribution would look something like this:
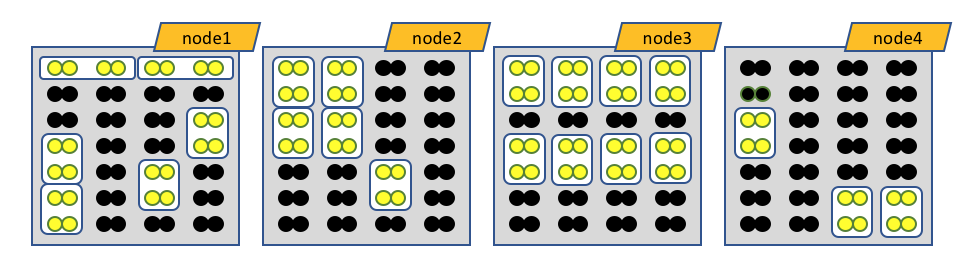
The white boxes represent MPI tasks, the yellow dots represent CPUs allocated to the MPI tasks, and the black dots are CPUs not allocated to the job. Because no constraints are placed on where tasks can be allocated via the --ntasks-per-node option, the MPI tasks distribute themselves wherever the slurm batch system finds room.
Obviously the number of MPI procs MUST equal --nodes times --ntasks-per-node. In this case 8 nodes, each with 4 MPI tasks per node, gives 32 MPI tasks total.
GPU-only:
In this case, because we are allocating all 4 GPUs on the gpu node with gpu:p100:4, it is probably best to allocate all the memory on the node as well, using --mem-per-cpu.
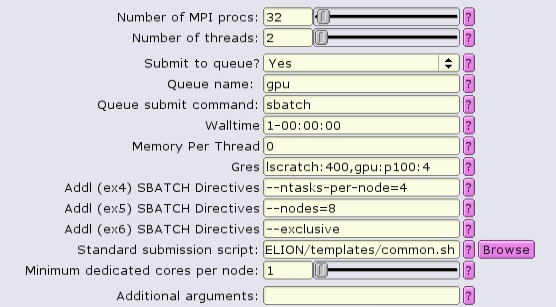
#!/bin/bash #SBATCH --ntasks=32 #SBATCH --partition=gpu #SBATCH --cpus-per-task=2 #SBATCH --error=run.err #SBATCH --output=run.out #SBATCH --time=1-00:00:00 #SBATCH --mem-per-cpu=0 #SBATCH --gres=lscratch:200,gpu:p100:4 #SBATCH --nodes=8 --ntasks-per-node=4 --exclusive ... RELION command here ...
A visual representation of this distribution would be:
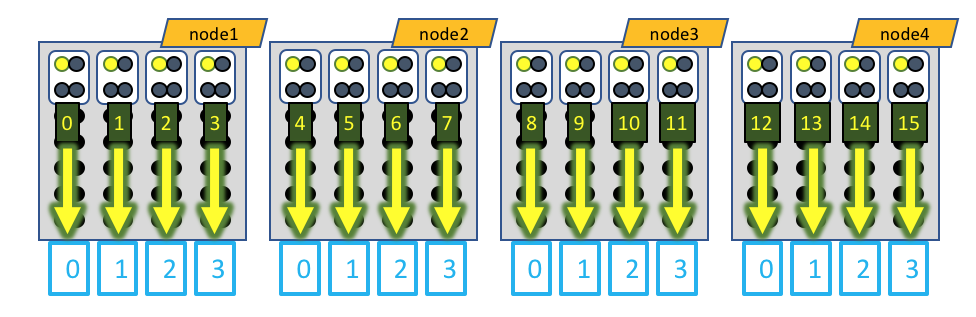
The white boxes represent MPI tasks, the yellow dots represent CPUs allocated to the MPI tasks, and the black dots are CPUs not allocated to the job. In this case, the GPU devices are represented by blue boxes, and each MPI task is explicitly mapped to a given GPU device. When running MotionCor2, only a single CPU of each MPI task actually generates load, so only a single CPU of the 4 allocated to each MPI task is active.
GPU-only:
For homogeneous+1 distribution, the total number of MPI procs is more than necessary, and must equal to the number of nodes times the number of tasks per node. --ntasks-per-node is set to 5, and --nodes is set to 8, so the total number of tasks is set to 40.
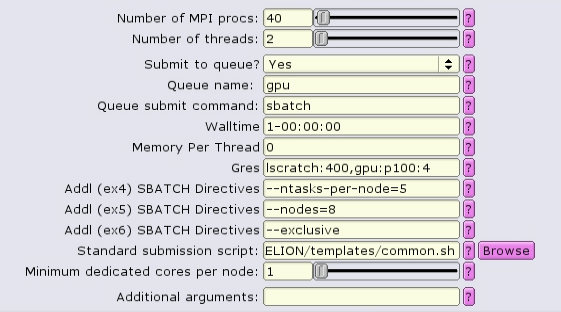
The batch script now contains a special source file, add_extra_MPI_task.sh, which creates the $SLURM_HOSTFILE and distributes the MPI tasks in an arbitrary fashion.
#!/bin/bash #SBATCH --ntasks=40 #SBATCH --partition=gpu #SBATCH --cpus-per-task=2 #SBATCH --error=run.err #SBATCH --output=run.out #SBATCH --time=1-00:00:00 #SBATCH --mem-per-cpu=0 #SBATCH --gres=lscratch:200,gpu:p100:4 #SBATCH --nodes=8 --ntasks-per-node=5 --exclusive source add_extra_MPI_task.sh ... RELION command here ...
Visually, this distribution would look something like this:
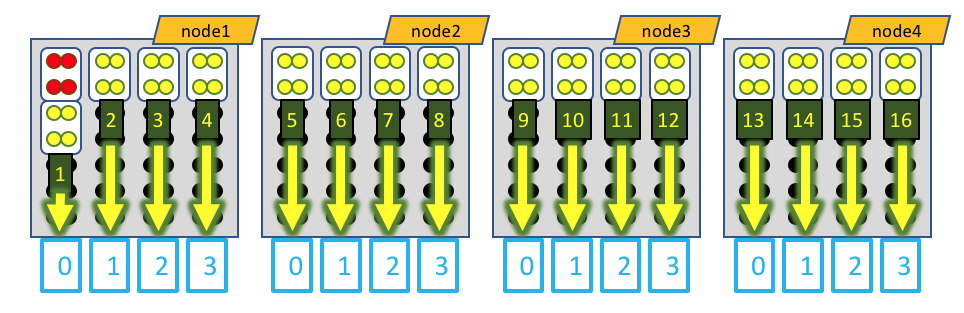
The white boxes represent MPI tasks, the yellow dots represent CPUs allocated to the MPI tasks, and the black dots are CPUs not allocated to the job. Again, the GPU devices are represented by blue boxes, and each MPI task is explicitly mapped to a given GPU device. However, in this case, the leader MPI task (as part of a RELION job) is allocated an MPI task, but does not do much and does not utilize a GPU device, so its CPUs are colored red.
Certain job-types (2D classification, 3D classification, and refinement) can benefit tremendously by using GPUs. Under the Compute tab, set 'Use GPU acceleration?' to 'Yes', and leave 'Which GPUs to use' blank:
The job must be configured to allocate GPUs. This can be done by setting input in the 'Running tab'.

For homogeneous distribution, the total number of MPI tasks should equal:
<number of GPUs per node> X <number of nodes>
and the value of --ntasks-per-node should be equal to <number of GPUs per node>
For homogeneous+1 distribution (requires --ntasks-per-node to be odd), the total number of MPI tasks should equal:
(<number of GPUs per node + 1>) X <number of nodes>
and the value of --ntasks-per-node should be equal to <number of GPUs per node + 1>
For more information about the GPUs available on the HPC/Biowulf cluster, see https://hpc.nih.gov/systems/.
By default, RELION comes supplied with a built-in motion correction tool:
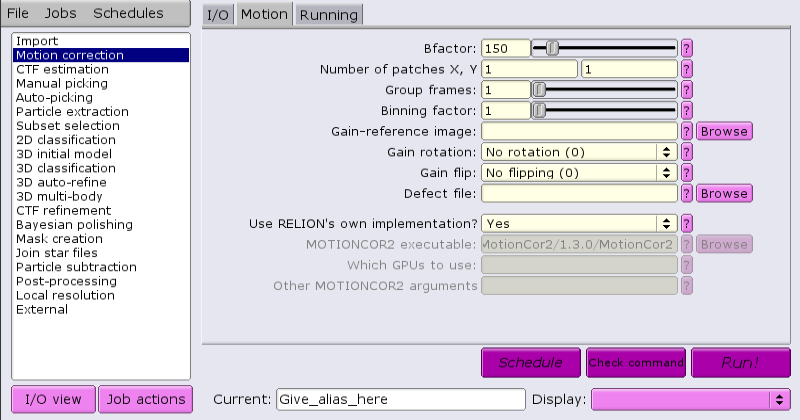
This tool does not require GPUs, but can run multi-threaded.
CPU-only motion correction can require a lot of memory, minimally 8g per cpu.
Each MPI process minimally needs:
width * height * (frame + 2 + X) * 4 bytes per MPI task.
where X is at least the number of threads per MPI process. For example, using relion_image_handler --stats --i to display the statistics of a in input .tif file,
049@EMPIAR-10204/Movies/20170630_3_00029_frameImage.tif : (x,y,z,n)= 3710 x 3838 x 1 x 1 ; avg= 1.0305 stddev= 1.01565 minval= 0 maxval= 75; angpix = 1
the MPI process would minimally require
3710 * 3838 * (49 + 2 + 1) * 4, or ~ 2.7 GB.
However, due to other factors, this can be multiplied by 4-10 times.
There is also an external application that can be used, MotionCor2 from Shawn Zheng of UCSF. This requires GPUs to run. Several steps must be done to ensure success. If running MotionCor2 within an interactive session, there must be at least one GPU allocated. Otherwise, GPUs must be allocated within a batch job from the GUI.
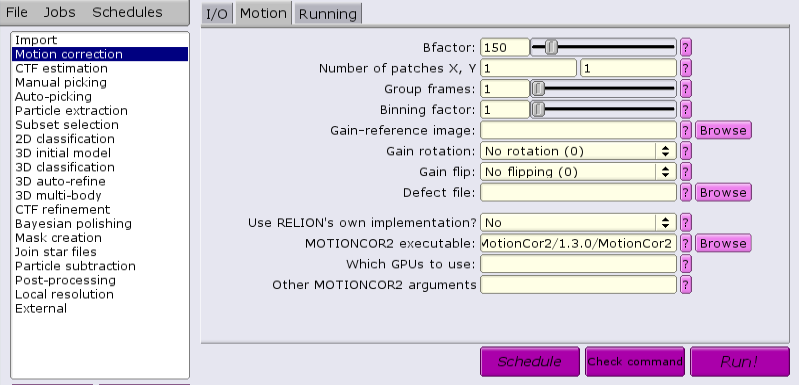
There are multiple applications and versions available for doing CTF estimation.
Under the CTFFIND-4.1 tab, change the answer to 'Use CTFFIND-4.1?' to 'Yes'.
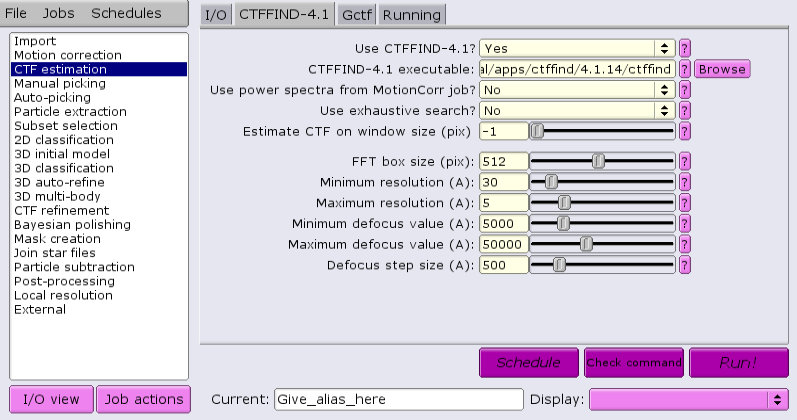
Under the CTFFIND-4.1 tab, change the answer to 'Use CTFFIND-4.1?' to 'No'.

Under the Gctf tab, change the answer to 'Use Gctf instead?' to 'Yes'. Keep in mind that GCTF requires GPUs.
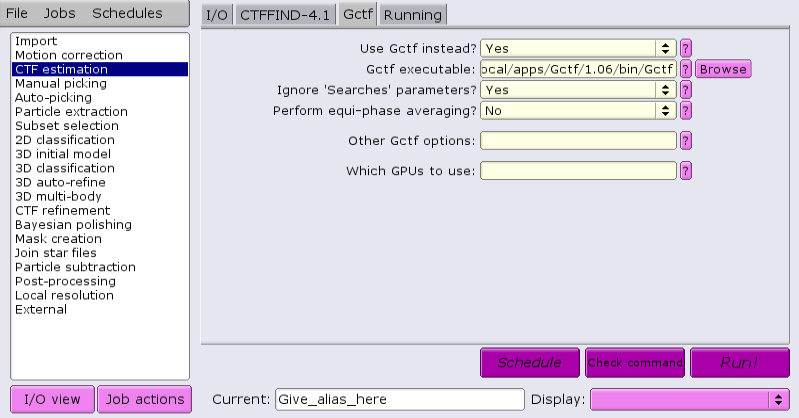
Topaz v0.2.5 is available for autopicking particles in RELION v4. Its location is determined by the environment variable RELION_TOPAZ_EXECUTABLE after loading the RELION module. It's path should appear in the "Topaz executable:" input box of the Topaz tab and is currently set to: /usr/local/apps/topaz/0.2.5/bin/topaz

PLEASE NOTE: Topaz can only run on a single gpu. Allocating more than one gpu to the job will cause the Topaz job to fail!
ERROR: AutoPicker::readTopazCoordinate
Topaz can run with multiple MPI processors. Here is an example of how to submit a Topaz job:
Topaz tab:
| Topaz executable | /path/to/topaz |
| Perform topaz picking? | No |
| Trained topaz model: | -- leave blank -- |
| Perform topaz training? | Yes |
| Particles STAR file for training: | /path/to/star |
Compute tab:
| Use GPU acceleration? | yes |
| Which GPUs to use: | -- leave blank -- |
Running tab:
| Number of MPI procs: | 1 |
| Number of threads: | 1 |
| Submit to queue? | yes |
| Queue name: | gpu |
| Walltime: | 24:00:00 |
| Memory Per Thread: | 20g |
| Gres: | gpu:p100:1 |
Topaz tab:
| Topaz executable | /path/to/topaz |
| Perform topaz picking? | Yes |
| Trained topaz model: | /path/to/model |
| Perform topaz training? | No |
| Particles STAR file for training: | -- leave blank -- |
Compute tab:
| Use GPU acceleration? | yes |
| Which GPUs to use: | -- leave blank -- |
Running tab:
| Number of MPI procs: | 4 |
| Number of threads: | 1 |
| Submit to queue? | yes |
| Queue name: | gpu |
| Walltime: | 24:00:00 |
| Memory Per Thread: | 20g |
| Gres: | gpu:p100:1 |
Long-running multi-node jobs can benefit from copying input data into local scratch space. The benefits stem from both increased I/O performance and the prevention of disruptions due to unforseen traffic on shared filesystems. Under the Compute tab, insert /lscratch/$SLURM_JOB_ID into the 'Copy particles to scratch directory' input:
Make sure that the total size of your particles can fit within the allocated local scratch space, as set in the 'Gres' input under the Running tab.

The batch script should contain the option --scratch_dir /lscratch/$SLURM_JOB_ID.
When running RELION on multiple CPUs, keep in mind both the partition (queue) and the nodes within that partition. Several of the partitions have subsets of nodetypes. Having a large RELION job running across different nodetypes may be detrimental. Specifically, cpu x6140 is not compatible with either one of the older cpus (x2680/x2695/x2630) on multinode. It's mandatory now to include --constraint in the "Additional SBATCH Directives" input so as to select a specific nodetype for RELION jobs on multinode. For example, --constraint x2680 would be a good choice for the multinode partition.
Please read https://hpc.nih.gov/policies/multinode.html for a discussion on making efficient use of multinode partition.
In benchmarking tests, RELION classification (2D & 3D) MPI jobs scale about the same as the number of CPUs increase, regardless of the combination of MPI procs and threads per MPI process. That is, a 3D classification job with 512 MPI procs and 2 threads per MPI proc runs about the same as with 128 MPI procs and 8 threads per MPI proc. Both utilize 1024 CPUs. However, refinement MPI jobs run dramatically faster with 16 threads per MPI proc than with 1.
This application requires a graphical connection.
Running RELION on the login node is not allowed. Please allocate an interactive node instead.
Additional sbatch options can be placed in the Additional SBATCH Directives: text boxes.

RELION allows additional options to be added to the command line as well:

While batch jobs running on the same node do not share CPUs, memory or local scratch space, they do share network interfaces to filesystems and other nodes (and can share GPUs by mistake). If one job on a node generates a heavy demand on these interfaces (e.g. performing lots of reads/writes to shared disk space, communicating a huge amount of packets between other nodes), then the other jobs on that node may suffer. To alleviate this, a job can be run with the --exclusive flag. It has been found that in general RELION jobs do best if run exclusively.
Typically, because GPU nodes are allocated with all GPUs on the node, the nodes are allocated exclusively by default, and there is no need to use --exclusive with GPU jobs. In fact, it adds quite a bit of complexity, and should be avoided.
The easiest way to run a CPU-only job exclusively is to use a special script template. There are three that are provided, one for each nodetype:
When using these templates, the only input parameters to be set are:
All other input parmeters will be ignored.
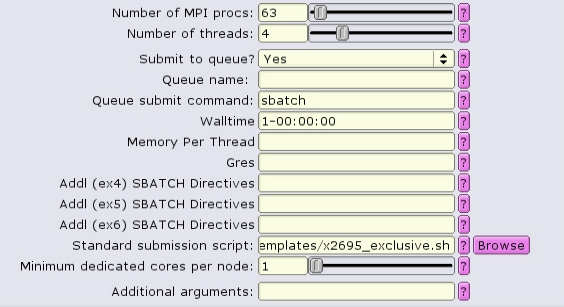
Alternatively when using the common template, exclusivity can be enabled by including --exclusive in the additional SBATCH directives boxes:

NOTE: --exclusive does not automatically allocate all the memory or lscratch on the node! Make sure that you designate the node resources needed, e.g.
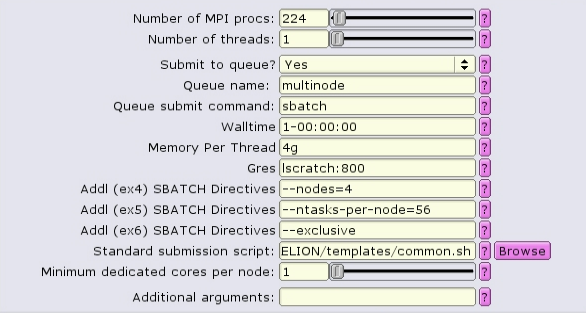
In this case, 4 nodes with 56 CPUs each are allocated. 4g of memory per CPU = 224Gb of RAM per node. To see resources available per node, type freen at the commandline.
Under certain circumstances, for example when the total size of input particles is small, pre-reading the particles into memory can improve performance. The amount of memory required depends on the number of particles (N) and the box_size:
N * box_size * box_size * 4 / (1024 * 1024 * 1024), in GB per MPI task.
Thus, 100,000 particles of box size 350 pixels would need ~43 GB of RAM per MPI task. This would reasonably fit on GPU nodes (240 GB) when running 17 tasks across 4 nodes, as the first node would have 5 MPI tasks for a total of 5*43, or 215 GB.
Under the Compute tab, change 'Pre-read all particles into RAM?' to 'Yes':

A few sample sets have been downloaded from https://www.ebi.ac.uk/pdbe/emdb/empiar/ for testing purposes. They are located here:
/fdb/app_testdata/cryoEM/
There are several known problems with RELION.
srun: error: cn1614: task 3: Exited with exit code 1Unfortunately, the leader MPI rank continues to run waiting for that rank to respond. It will wait forever until the slurm job times out. If you see your job running forever without progression, you should cancel the job and either start over or continue from where the classification left off.
ERROR: out of memory in /usr/local/apps/RELION/git/2.1.0/src/gpu_utils/cuda_mem_utils.h at line 576 (error-code 2) [cn4174:27966] *** Process received signal *** [cn4174:27966] Signal: Segmentation fault (11)At best, you can limit the number of classes, pool size, or box size to avoid this. But, you may need to run on CPUs only. See here for a listing of the GPU nodes and their properties, specifically VRAM.
[cn4021:mpi_rank_198][handle_cqe] Send desc error in msg to 196, wc_opcode=0 [cn4021:mpi_rank_198][handle_cqe] Msg from 196: wc.status=12, wc.wr_id=0xc1cba80, wc.opcode=0, vbuf->phead->type=0 = MPIDI_CH3_PKT_EAGER_SEND [cn4021:mpi_rank_198][handle_cqe] src/mpid/ch3/channels/mrail/src/gen2/ibv_channel_manager.c:547: [] Got completion with error 12, vendor code=0x81, dest rank=196 : No such file or directory (2)Rerun the job, this time allocating as much /lscratch space as possible.
terminate called after throwing an instance of 'std::bad_alloc' what(): std::bad_alloc [cn3672:mpi_rank_0][error_sighandler] Caught error: Aborted (signal 6) srun: error: cn3672: task 0: AbortedLikely the AutoPick step completed, and the output coordinates are available in the output job directory. Permanently fix the problem by locating the corrupt or missing micrograph and remove it using Select or Import. This can be done using the relion_image_handler --stats command or by simply comparing the sizes of the micrograph files and finding the ones that don't belong.
ERROR: CudaCustomAllocator out of memory [requestedSpace: 1672740864 B] [largestContinuousFreeSpace: 1409897472 B] [totalFreeSpace: 1577153024 B]This occurs when either the number of MPI tasks exceeds the number of GPUs available, or then number of threads is greater than is reasonable for the gpu type.
ERROR in removing non-dose weighted image: MotionCorr/job005/Movies/20170630_3_00443_frameImage.mrcLooking more closely at the .err files in the Movies subdirectory (MotionCorr/job005/Movies/20170630_3_00443_frameImage.err):
Error: All GPUs are in use, quit.Only assign a single thread per GPU. This occurs when either the number of MPI tasks exceeds the number of GPUs available, or then number of threads is unreasonable for the gpu type.
WARNING: trying to read pipeline.star, but directory .relion_lock exists.Hunt down and stop all instances of the RELION GUI, then remove the lock directory if it still exists:
rm -rf .relion_lock
ERROR: PipeLine::read: cannot find name or type in pipeline_nodes tableCopying a default_pipeline.star file from the latest run job into the project directory should fix this.
ERROR: FFTW plans cannot be createdLikely one or more of the input micrographs is empty or corrupted, possibly from data transfer errors. Have a look at the micrographs, looking for those whose sizes are abnormal:
ls -l *.mrc ... -rw-r-----. 1 user user 1342214144 Sep 15 03:08 Data_9999999_9999999_000021.mrc -rw-r-----. 1 user user 1342214144 Sep 14 23:55 Data_9999999_9999999_000022.mrc -rw-r-----. 1 user user 0 Sep 15 00:20 Data_9999999_9999999_000023.mrc -rw-r-----. 1 user user 1342214144 Sep 14 23:51 Data_9999999_9999999_000024.mrc -rw-r-----. 1 user user 555008000 Sep 14 14:29 Data_9999999_9999999_000025.mrc -rw-r-----. 1 user user 1342214144 Sep 14 16:22 Data_9999999_9999999_000026.mrc -rw-r-----. 1 user user 1342214144 Sep 14 22:53 Data_9999999_9999999_000027.mrc ...or run the relion_image_handler --stats command on each file (this can take quite a while).