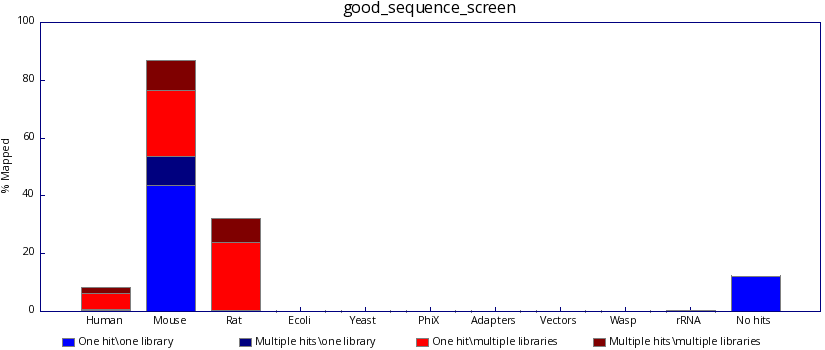
fastq_screen generates the following graphs demonstrating the proportion of your library was able to map:
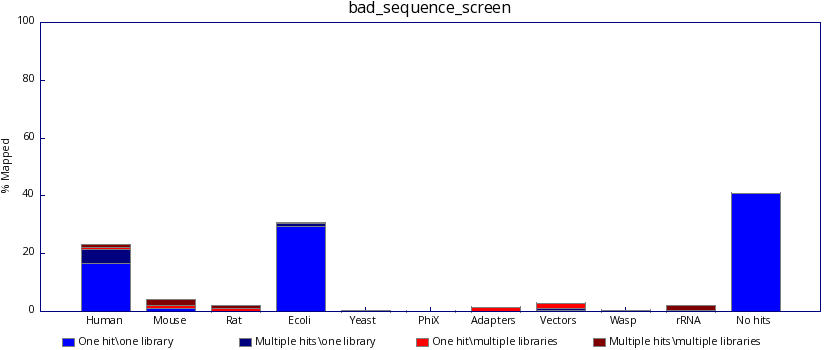
In contrast, poor sequencing results will include results from one or more unexpected species.
A tool for multi-genome mapping and quality control. It allows you to screen a library of sequences in FastQ format against a set of sequence databases so you can see if the composition of the library matches with what you expect.
Features
fastq_screen generates the following graphs demonstrating the proportion of your library was able to map:
In contrast, poor sequencing results will include results from one or more unexpected species.
Allocate an interactive session and run the program.
Sample session (user input in bold):
[user@biowulf]$ sinteractive --cpus-per-task=8 salloc.exe: Pending job allocation 46116226 salloc.exe: job 46116226 queued and waiting for resources salloc.exe: job 46116226 has been allocated resources salloc.exe: Granted job allocation 46116226 salloc.exe: Waiting for resource configuration salloc.exe: Nodes cn3144 are ready for job [user@cn3144 ~]$ module load fastq_screen [user@cn3144 ~]$ mkdir /data/$USER/fastq_screen [user@cn3144 ~]$ cd /data/$USER/fastq_screen [user@cn3144 ~]$ cp $FASTQ_SCREEN_TEST_DATA/* . [user@cn3144 ~]$ fastq_screen --tag fqs_test_dataset.fastq.gz Using fastq_screen v0.14.0 Reading configuration from '/usr/local/apps/fastq_screen/0.14.0/bin/fastq_screen_v0.14.0/fastq_screen.conf' Aligner (--aligner) not specified, but Bowtie2 path and index files found: mapping with Bowtie2 Using '/usr/local/apps/bowtie/2-2.4.1/bin/bowtie2' as Bowtie 2 path Adding database Human Adding database Mouse Adding database Rat Adding database Drosophila Adding database Worm Adding database Yeast Adding database Arabidopsis Adding database Ecoli Adding database rRNA Adding database MT Adding database PhiX Adding database Lambda Adding database Vectors Adding database Adapters Using 8 threads for searches Option --subset set to 0: processing all reads in FASTQ files Processing fqs_test_dataset.fastq.gz Not making data subset Searching fqs_test_dataset.fastq.gz_temp_subset.fastq against Human Searching fqs_test_dataset.fastq.gz_temp_subset.fastq against Mouse Searching fqs_test_dataset.fastq.gz_temp_subset.fastq against Rat Searching fqs_test_dataset.fastq.gz_temp_subset.fastq against Drosophila Searching fqs_test_dataset.fastq.gz_temp_subset.fastq against Worm Searching fqs_test_dataset.fastq.gz_temp_subset.fastq against Yeast Searching fqs_test_dataset.fastq.gz_temp_subset.fastq against Arabidopsis Searching fqs_test_dataset.fastq.gz_temp_subset.fastq against Ecoli Searching fqs_test_dataset.fastq.gz_temp_subset.fastq against rRNA Searching fqs_test_dataset.fastq.gz_temp_subset.fastq against MT Searching fqs_test_dataset.fastq.gz_temp_subset.fastq against PhiX Searching fqs_test_dataset.fastq.gz_temp_subset.fastq against Lambda Searching fqs_test_dataset.fastq.gz_temp_subset.fastq against Vectors Searching fqs_test_dataset.fastq.gz_temp_subset.fastq against Adapters Processing complete [user@cn3144 ~]$ exit salloc.exe: Relinquishing job allocation 46116226 [user@biowulf ~]$
Create a batch input file (e.g. fastq_screen.sh). For example:
#!/bin/bash set -e module load fastq_screen fastq_screen --tag fqs_test_dataset.fastq.gz
Submit this job using the Slurm sbatch command.
sbatch --cpus-per-task=8 --mem=2g fastq_screen.sh
Create a swarmfile (e.g. fastq_screen.swarm). For example:
cd dir1;fastq_screen --tag R1.fq cd dir2;fastq_screen --tag R2.fq cd dir3;fastq_screen --tag R3.fq cd dir4;fastq_screen --tag R4.fq
Submit this job using the swarm command.
swarm -f fastq_screen.swarm [-g #] [-t 8] --module fastq_screenwhere
| -g # | Number of Gigabytes of memory required for each process (1 line in the swarm command file) |
| -t # | Number of threads/CPUs required for each process (1 line in the swarm command file). |
| --module fastq_screen | Loads the fastq_screen module for each subjob in the swarm |