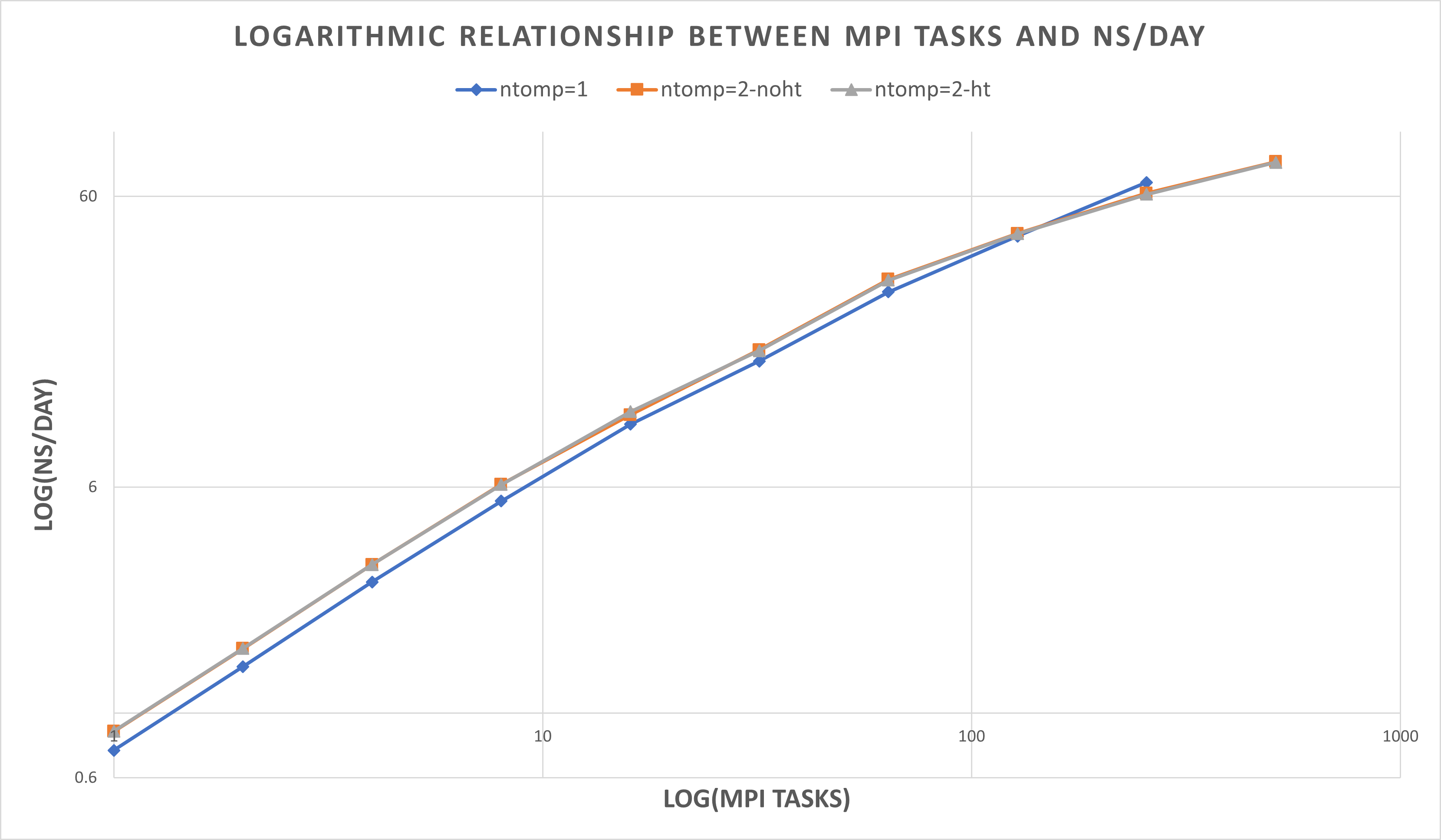
| ntomp = 1 | blue line | 1 thread per MPI task, 1 MPI task per physical core. No hyperthreading |
| ntomp = 2-noht | orange line | 2 threads per MPI task, no hyperthreading. This uses twice as many cores as the options above, but with threads per core = 1. |
| ntomp = 2-ht | grey line | 2 threads per MPI task, hyperthreading enabled. This effectively uses the same number of cores as ntomp=1, but uses both hyperthreaded CPUs on each core. |
| # MPI tasks | ntomp = 1 ns/day | ntomp = 2-noht ns/day | ntomp = 2-ht ns/day 1 | 0.745 | 0.866 | 0.867
| 2 | 1.448 | 1.669 | 1.672
| 4 | 2.828 | 3.244 | 3.244
| 8 | 5.372 | 6.132 | 6.121
| 16 | 9.861 | 10.63 | 10.909
| 32 | 16.28 | 17.803 | 17.666
| 64 | 28.164 | 31.117 | 30.872
| 128 | 43.785 | 44.728 | 44.651
| 256 | 67.001 | 61.482 | 61.008
| 512 | 87.604 | 78.883 | 78.601
| |
Larger numbers of MPI tasks should use ntomp = 2, all MPI tasks run most effectively assigning ntasks-per-core = 1.
[Jan 2023] The ADH benchmark suite is available on the Gromacs website. The tests below used the ADH_cubic benchmark..
For running gromacs on GPU: Running mdrun with GPUs.
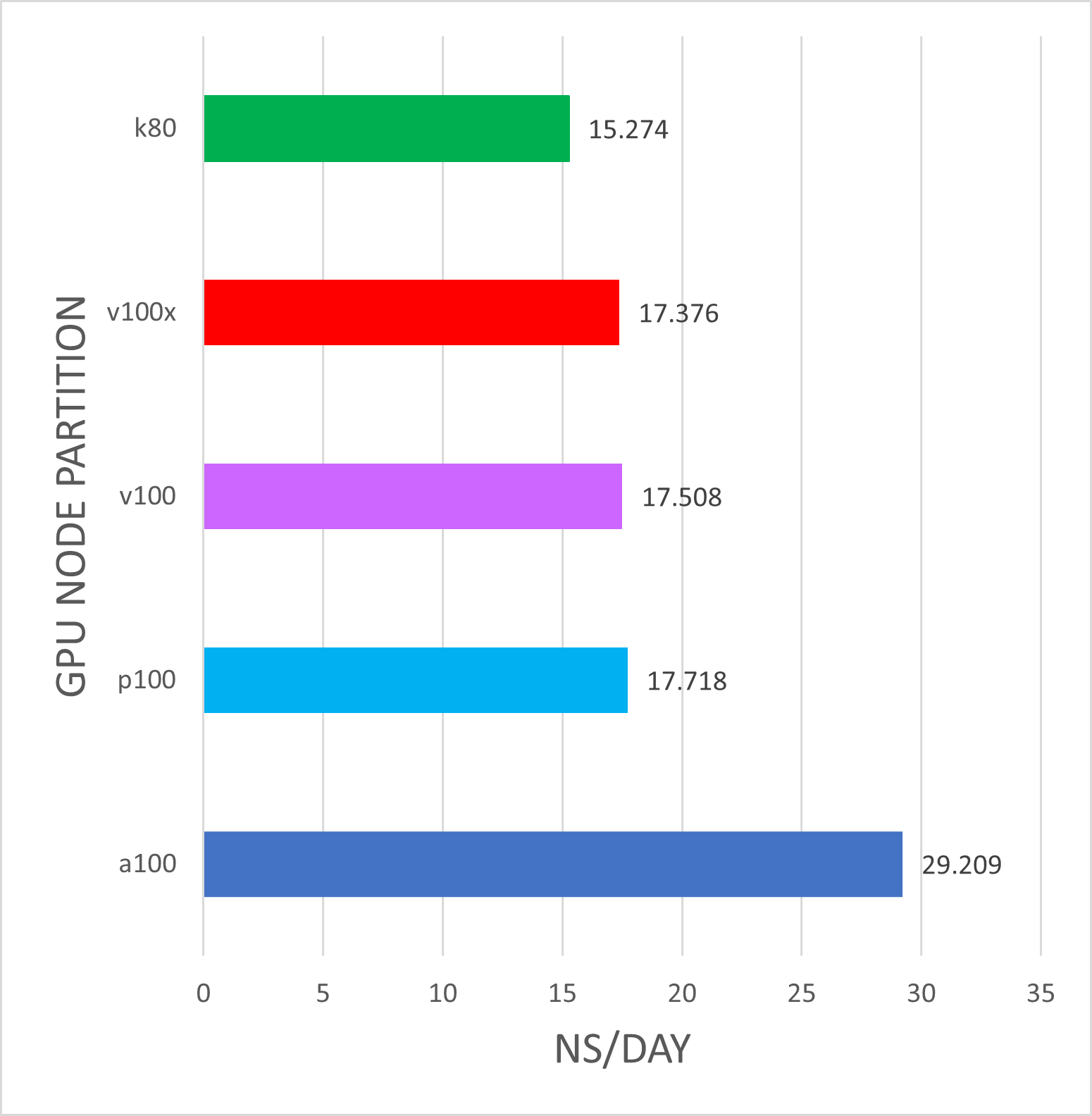
v100's are Biowulf phase3 nodes Intel(R) Xeon(R) CPU E5-2680 v4 @ 2.40GHz.
v100x's are Biowulf phase5 nodes Intel Gold 6140 @ 2.30GHz.
p100's are Biowulf phase4 nodes Intel(R) Xeon(R) CPU E5-2680 v4 @ 2.40GHz.
a100s's are Biowulf phase6 nodes AMD EPYC 7543P 32-Core Processor.
| For running GPU jobs, please use the freen command to determine which GPU paritions are available. For running these jobs, an appopriate submission command would be:
sbatch --partition=gpu \ --gres=gpu:xxx:1 --ntasks=2 \ --ntasks-per-core=1 --exclusive jobscript For this benchmark, there is a significant advantage to running Gromacs a100 nodes over k80 nodes. There is typically no advantage to running on more than a single GPU device. Single-GPU jobs were run on a single GPU with ntasks=2, n-tasks-per-core=1, and ntomp=2. These results may be different for larger molecular systems, so please run your own benchmarks to verify that your jobs benefit from the GPUs (and send us any interesting results!)
|