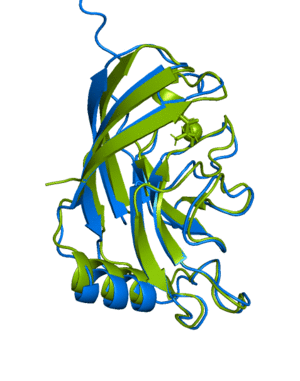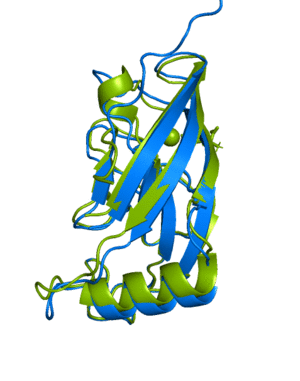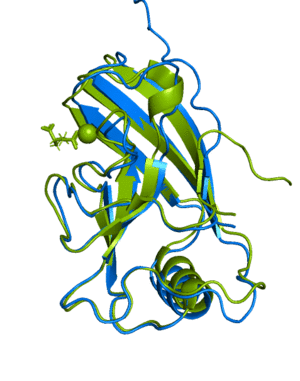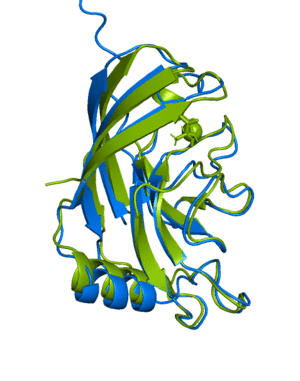
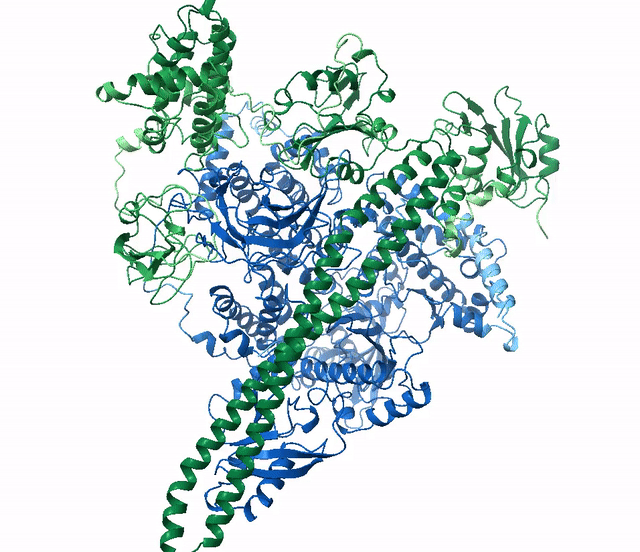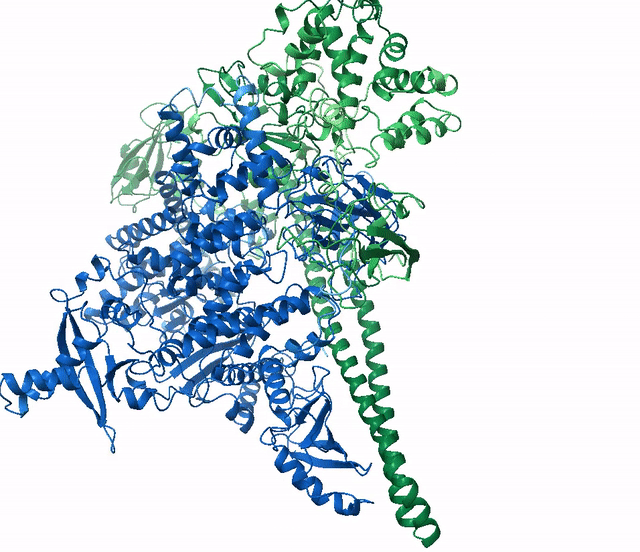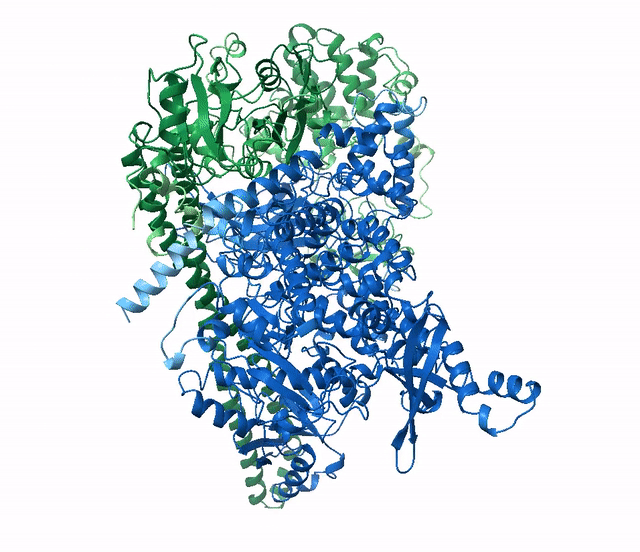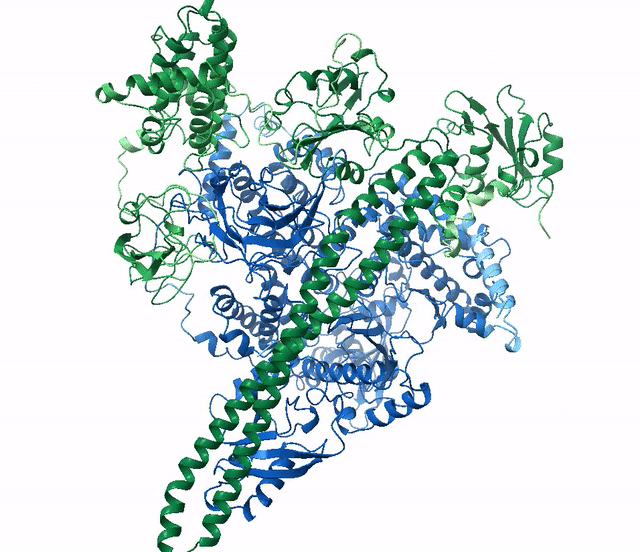
ranked_0.pdb, blue) aligned with the actual
structure for this protein
(6Y4F, green)From the official documentation
This package provides an implementation of the inference pipeline of AlphaFold v2.0. This is a completely new model that was entered in CASP14 and published in Nature. For simplicity, we refer to this model as AlphaFold throughout the rest of this document.
Please do not run more than 500 AlphaFold2 Multiple Sequence Alignment (MSA) jobs concurrently, as this may degrade system performance. For large-scale predictions, please use ColabFold for MSA and contact the HPC staff at staff@hpc.nih.gov
Please also check our general guidance on alphafold-related applications.
Code availability: AlphaFold 3 will be available as a non-commercial usage only server at https://www.alphafoldserver.com, with restrictions on allowed ligands and covalent modifications. Pseudocode describing the algorithms is available in the Supplementary Information. Code is not provided.
relax and afpdb2cif scripts to alphafold/2.3.2
modulealphafold
command was added as an alias to the awkwardly named run_singularity for.--run_relax has been replaced with --models_to_relax with a default
of best. That means that only the best model (ranked_0.pdb) will be relaxed.jax python module is no loger required to read the pickled results files--hhblits_extra_opts option was ported from msa to run_singularityrun_singularity --hhblits_extra_opts="-maxres 80000 -prepre_smax_thresh 50" ...msa utility script has been disabledmsa script may have been implicated in file system problems.
The script has been removed until futher notice.--is_prokaryote_list option from run_singularity
and msa as the prokaryotic pairing algorithm did not actually improve
accuracy on average--num_multimer_predictions_per_model=N option to
run_singularity. Runs N predictions per multimer model - each
with different seeds. Defaults to 5 and will increase runtime--model_config option to run_singularity. This
allows users to use a customized alphafold/model/config.py to alphafold
for tuning certain parameters--[no]run_relax and --[no]enable_gpu_relaxmsa). This is
highly recommended as MSA generation consumes
about 60% of runtime and does not make use of GPU. The workflow is to run msa generation on CPU nodes
and then run model prediction on a GPU node with run_singularity --use_precomputed_msas ....
This script also supports adding extra options to hhblits or overriding some alphafold defaults with option
--hhblits_extra_opts.
Note that this script cannot run on x2650 nodes because it depends on an AVX2 hhblits build.--preset option was renamed to --model_preset--db_preset=reduced_dbs option is now supported for all alphafold versions--use_ptm option became obsolete with introduction of the official --model_preset
option and was removed--use_ptm option to run_singularity$ALPHAFOLD2_TEST_DATA/fdb/alphafold2/It's recommended to run AlphaFold with sbatch jobs to allow for longer walltime, particularly during model inference. This section is about examples of interactive jobs, which are primarily intended for testing, troubleshooting and developing AlphaFold job-related scripts. Allocate an interactive session and run the program. We strongly recommend dividing the AlphaFold pipeline into two steps: performing CPU-based multiple sequence alignment (MSA) on a CPU node, followed by GPU-based model prediction on a GPU node.
### To run MSA on CPU: ### [user@biowulf]$ sinteractive --mem=60g --cpus-per-task=8 --gres=lscratch:100 salloc.exe: Pending job allocation 46116226 salloc.exe: job 46116226 queued and waiting for resources salloc.exe: job 46116226 has been allocated resources salloc.exe: Granted job allocation 46116226 salloc.exe: Waiting for resource configuration salloc.exe: Nodes cn3144 are ready for job [user@cn3144]$ module load alphafold2/2.3.2
To predict the structure of a protein already in PDB without using its
experimental structure as a template set max_template_date to
before the release date of the structure. For example, to reproduce the T1049
CASP14 target with 144 aa. On a V100x this prediction runs for about 1h.
[user@cn3144]$ run_singularity --helpfull # use --help for shorter help message
Singularity launch script for Alphafold.
flags:
/usr/local/apps/alphafold2/2.3.2/bin/run_singularity:
--[no]benchmark: Run multiple JAX model evaluations to obtain a timing that excludes the compilation time,
which should be more indicative of the time required for inferencing many proteins.
(default: 'false')
--db_preset: <full_dbs|reduced_dbs>: Choose preset MSA database configuration - smaller genetic
database config (reduced_dbs) or full genetic database config (full_dbs)
(default: 'full_dbs')
--[no]dry_run: Print command that would have been executed and exit.
(default: 'false')
--[no]enable_gpu_relax: Run relax on GPU if GPU is enabled.
(default: 'true')
--fasta_paths: Paths to FASTA files, each containing a prediction target that will be folded one after
another. If a FASTA file contains multiple sequences, then it will be folded as a multimer. Paths should
be separated by commas. All FASTA paths must have a unique basename as the basename is used to name the
output directories for each prediction. (a comma separated list)
--gpu_devices: Comma separated list of devices to pass to NVIDIA_VISIBLE_DEVICES.
(default: 'all')
--max_template_date: Maximum template release date to consider (ISO-8601 format: YYYY-MM-DD). Important
if folding historical test sets.
--model_config: Use this file instead of default alphafold/model/config.py
--model_preset: <monomer|monomer_casp14|monomer_ptm|multimer>: Choose preset model configuration -
the monomer model, the monomer model with extra ensembling, monomer model with pTM head, or multimer model
(default: 'monomer')
--num_multimer_predictions_per_model: How many predictions (each with a different random seed) will be
generated per model. E.g. if this is 2 and there are 5 models then there will be 10 predictions per
input. Note: this FLAG only applies if model_preset=multimer
(default: '5')
(an integer)
--output_dir: Path to a directory that will store the results.
--[no]run_relax: Whether to run the final relaxation step on the predicted models. Turning relax off might
result in predictions with distracting stereochemical violations but might help in case you are having
issues with the relaxation stage.
(default: 'true')
--[no]use_gpu: Enable NVIDIA runtime to run with GPUs.
(default: 'true')
--[no]use_precomputed_msas: Whether to read MSAs that have been written to disk instead of running the
MSA tools. The MSA files are looked up in the output directory, so it must stay the same between multiple
runs that are to reuse the MSAs. WARNING: This will not check if the sequence, database or configuration
have changed.
(default: 'false')
...
absl.logging:
--[no]alsologtostderr: also log to stderr?
(default: 'false')
--log_dir: directory to write logfiles into
(default: '')
--logger_levels: Specify log level of loggers. The format is a CSV list of `name:level`. Where `name` is the
logger name used with `logging.getLogger()`, and `level` is a level name (INFO, DEBUG, etc). e.g.
`myapp.foo:INFO,other.logger:DEBUG`
(default: '')
--[no]logtostderr: Should only log to stderr?
(default: 'false')
--[no]showprefixforinfo: If False, do not prepend prefix to info messages when it's logged to stderr, --verbosity
is set to INFO level, and python logging is used.
(default: 'true')
--stderrthreshold: log messages at this level, or more severe, to stderr in addition to the logfile.
Possible values are 'debug', 'info', 'warning', 'error', and 'fatal'. Obsoletes --alsologtostderr.
Using --alsologtostderr cancels the effect of this flag. Please also note that this flag is
subject to --verbosity and requires logfile not be stderr.
(default: 'fatal')
-v,--verbosity: Logging verbosity level. Messages logged at this level or lower will be included. Set to 1
for debug logging. If the flag was not set or supplied, the value will be changed from the default of
-1 (warning) to 0 (info) after flags are parsed.
(default: '-1')
(an integer)
...
[user@cn3144]$ run_singularity \
--model_preset=monomer \
--fasta_paths=$ALPHAFOLD2_TEST_DATA/T1049.fasta \
--max_template_date=2022-12-31 \
--msas_only \
--output_dir=$PWD
###
### or use the equivalent alphafold alias
###
[user@cn3144]$ alphafold \
--model_preset=monomer \
--fasta_paths=$ALPHAFOLD2_TEST_DATA/T1049.fasta \
--max_template_date=2022-12-31 \
--msas_only \
--output_dir=$PWD
###
### Then run model inference on GPU:
###
[user@biowulf]$ sinteractive --mem=60g --cpus-per-task=8 --gres=lscratch:100,gpu:v100x:1
salloc.exe: Pending job allocation 46116226
salloc.exe: job 46116226 queued and waiting for resources
salloc.exe: job 46116226 has been allocated resources
salloc.exe: Granted job allocation 46116226
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn3144 are ready for job
[user@cn3144]$ module load alphafold2/2.3.2
[user@cn3144]$ alphafold \
--model_preset=monomer \
--fasta_paths=$ALPHAFOLD2_TEST_DATA/T1049.fasta \
--max_template_date=2022-12-31 \
--use_precomputed_msas \
--output_dir=$PWD
[user@cn3144]$ tree T1049
T1049/
├── [user 1.1M] features.pkl
├── [user 4.0K] msas
│ ├── [user 33K] bfd_uniclust_hits.a3m
│ ├── [user 18K] mgnify_hits.sto
│ └── [user 121K] uniref90_hits.sto
├── [user 170K] ranked_0.pdb # <-- shown below
├── [user 170K] ranked_1.pdb
├── [user 170K] ranked_2.pdb
├── [user 171K] ranked_3.pdb
├── [user 170K] ranked_4.pdb
├── [user 330] ranking_debug.json
├── [user 170K] relaxed_model_1.pdb
├── [user 170K] relaxed_model_2.pdb
├── [user 170K] relaxed_model_3.pdb
├── [user 170K] relaxed_model_4.pdb
├── [user 171K] relaxed_model_5.pdb
├── [user 11M] result_model_1.pkl
├── [user 11M] result_model_2.pkl
├── [user 11M] result_model_3.pkl
├── [user 11M] result_model_4.pkl
├── [user 11M] result_model_5.pkl
├── [user 771] timings.json
├── [user 87K] unrelaxed_model_1.pdb
├── [user 87K] unrelaxed_model_2.pdb
├── [user 87K] unrelaxed_model_3.pdb
├── [user 87K] unrelaxed_model_4.pdb
└── [user 87K] unrelaxed_model_5.pdb
The processes prior to model inference on the GPU consumed up to 40 GB of memory for this protein. Memory requirements will vary with different size proteins.
ranked_0.pdb, blue) aligned with the actual
structure for this protein
(6Y4F, green)The next example shows how to run a multimer model (available from version 2.1.1). The example used is a recently published PI3K structure.
[user@biowulf]$ sinteractive --mem=60g --cpus-per-task=8 --gres=lscratch:100
[user@cn3144]$ module load alphafold2/2.3.2
[user@cn3144]$ cat $ALPHAFOLD2_TEST_DATA/pi3k.fa
>sp|P27986|P85A_HUMAN Phosphatidylinositol 3-kinase regulatory subunit alpha OS=Homo sapiens OX=9606 GN=PIK3R1 PE=1 SV=2
MSAEGYQYRALYDYKKEREEDIDLHLGDILTVNKGSLVALGFSDGQEARPEEIGWLNGYN
ETTGERGDFPGTYVEYIGRKKISPPTPKPRPPRPLPVAPGSSKTEADVEQQALTLPDLAE
QFAPPDIAPPLLIKLVEAIEKKGLECSTLYRTQSSSNLAELRQLLDCDTPSVDLEMIDVH
VLADAFKRYLLDLPNPVIPAAVYSEMISLAPEVQSSEEYIQLLKKLIRSPSIPHQYWLTL
QYLLKHFFKLSQTSSKNLLNARVLSEIFSPMLFRFSAASSDNTENLIKVIEILISTEWNE
RQPAPALPPKPPKPTTVANNGMNNNMSLQDAEWYWGDISREEVNEKLRDTADGTFLVRDA
STKMHGDYTLTLRKGGNNKLIKIFHRDGKYGFSDPLTFSSVVELINHYRNESLAQYNPKL
DVKLLYPVSKYQQDQVVKEDNIEAVGKKLHEYNTQFQEKSREYDRLYEEYTRTSQEIQMK
RTAIEAFNETIKIFEEQCQTQERYSKEYIEKFKREGNEKEIQRIMHNYDKLKSRISEIID
SRRRLEEDLKKQAAEYREIDKRMNSIKPDLIQLRKTRDQYLMWLTQKGVRQKKLNEWLGN
ENTEDQYSLVEDDEDLPHHDEKTWNVGSSNRNKAENLLRGKRDGTFLVRESSKQGCYACS
VVVDGEVKHCVINKTATGYGFAEPYNLYSSLKELVLHYQHTSLVQHNDSLNVTLAYPVYA
QQRR
>sp|P42336|PK3CA_HUMAN Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha isoform OS=Homo sapiens OX=9606 GN=PIK3CA PE=1 SV=2
MPPRPSSGELWGIHLMPPRILVECLLPNGMIVTLECLREATLITIKHELFKEARKYPLHQ
LLQDESSYIFVSVTQEAEREEFFDETRRLCDLRLFQPFLKVIEPVGNREEKILNREIGFA
IGMPVCEFDMVKDPEVQDFRRNILNVCKEAVDLRDLNSPHSRAMYVYPPNVESSPELPKH
IYNKLDKGQIIVVIWVIVSPNNDKQKYTLKINHDCVPEQVIAEAIRKKTRSMLLSSEQLK
LCVLEYQGKYILKVCGCDEYFLEKYPLSQYKYIRSCIMLGRMPNLMLMAKESLYSQLPMD
CFTMPSYSRRISTATPYMNGETSTKSLWVINSALRIKILCATYVNVNIRDIDKIYVRTGI
YHGGEPLCDNVNTQRVPCSNPRWNEWLNYDIYIPDLPRAARLCLSICSVKGRKGAKEEHC
PLAWGNINLFDYTDTLVSGKMALNLWPVPHGLEDLLNPIGVTGSNPNKETPCLELEFDWF
SSVVKFPDMSVIEEHANWSVSREAGFSYSHAGLSNRLARDNELRENDKEQLKAISTRDPL
SEITEQEKDFLWSHRHYCVTIPEILPKLLLSVKWNSRDEVAQMYCLVKDWPPIKPEQAME
LLDCNYPDPMVRGFAVRCLEKYLTDDKLSQYLIQLVQVLKYEQYLDNLLVRFLLKKALTN
QRIGHFFFWHLKSEMHNKTVSQRFGLLLESYCRACGMYLKHLNRQVEAMEKLINLTDILK
QEKKDETQKVQMKFLVEQMRRPDFMDALQGFLSPLNPAHQLGNLRLEECRIMSSAKRPLW
LNWENPDIMSELLFQNNEIIFKNGDDLRQDMLTLQIIRIMENIWQNQGLDLRMLPYGCLS
IGDCVGLIEVVRNSHTIMQIQCKGGLKGALQFNSHTLHQWLKDKNKGEIYDAAIDLFTRS
CAGYCVATFILGIGDRHNSNIMVKDDGQLFHIDFGHFLDHKKKKFGYKRERVPFVLTQDF
LIVISKGAQECTKTREFERFQEMCYKAYLAIRQHANLFINLFSMMLGSGMPELQSFDDIA
YIRKTLALDKTEQEALEYFMKQMNDAHHGGWTTKMDWIFHTIKQHALN
[user@cn3144]$ alphafold \
--fasta_paths=$ALPHAFOLD2_TEST_DATA/pi3k.fa \
--max_template_date=2021-11-01 \
--model_preset multimer \
--msas_only \
--output_dir=$PWD
...snip...
[user@cn3144]$ exit
[user@biowulf]$ sinteractive --mem=60g --cpus-per-task=8 --gres=lscratch:100,gpu:v100x:1
[user@cn3144]$ module load alphafold2/2.3.2
[user@cn3144]$ alphafold \
--fasta_paths=$ALPHAFOLD2_TEST_DATA/pi3k.fa \
--max_template_date=2021-11-01 \
--model_preset multimer \
--use_precomputed_msas \
--num_multimer_predictions_per_model=2 \
--output_dir=$PWD
...snip...
[user@cn3144]$ exit
The model .pkl files which, unlike the .pdb files, are not re-ordered into ranked_ files, contain a lot of information about the models. These are python pickle files and python can be used to explore and visualize them. For example:
[user@cn3144]$ conda activate my_py39 # needs jupyter and the packages imported below
[user@cn3144]$ cd T1049
[user@cn3144]$ jupyter console
In [1]: import pickle
In [2]: import json
In [3]: import pprint
In [4]: import jax # only needed for version 2.3.1
In [5]: pprint.pprint(json.load(open("ranking_debug.json", encoding="ascii")))
{'order': ['model_2_pred_0',
'model_3_pred_0',
'model_1_pred_0',
'model_5_pred_0',
'model_4_pred_0'],
'plddts': {'model_1_pred_0': 88.44386138278787,
'model_2_pred_0': 91.83564104655056,
'model_3_pred_0': 88.49961929441032,
'model_4_pred_0': 86.73066329994059,
'model_5_pred_0': 87.4009420322368}}
### so model 2 is the best model in this run and corresponds to ranked_0.pdf
In [6]: best_model = pickle.load(open("result_model_2_pred_0.pkl", "rb"))
In [7]: list(best_model.keys())
Out[7]:
['distogram',
'experimentally_resolved',
'masked_msa',
'predicted_lddt',
'structure_module',
'plddt',
'ranking_confidence']
In [8]: best_model['plddt'].shape
Out[8]: (141,)
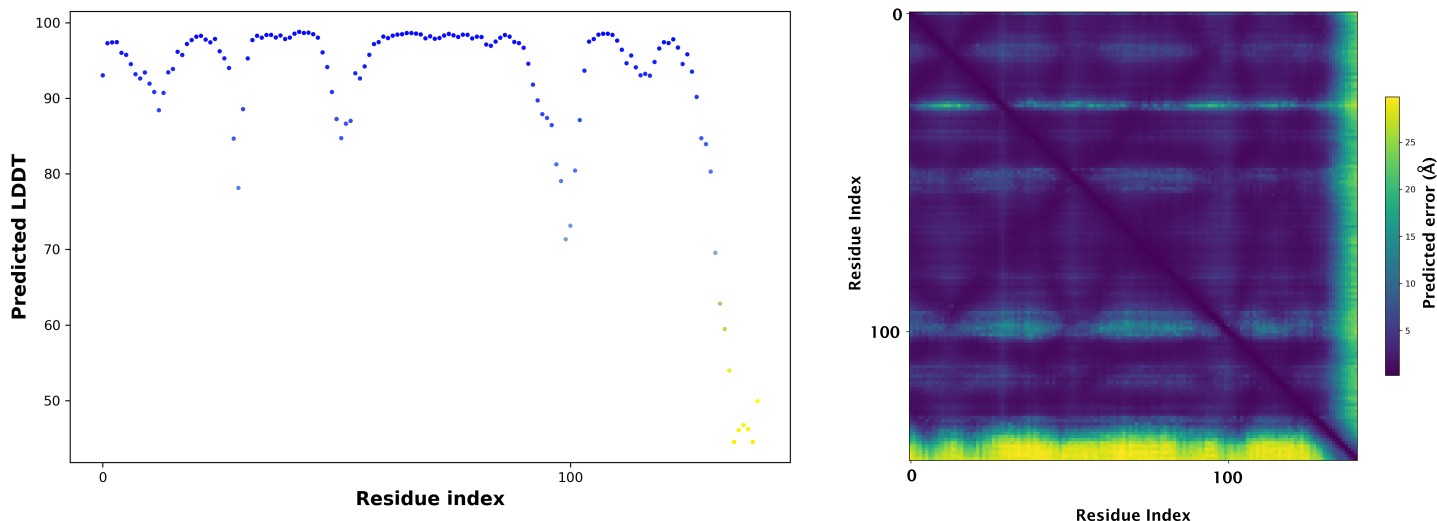
The predicted alignment error (PAE) is only produced by the monomer_ptm and
multimer models. Since version 2.2.0 we also include alphapickle
with alphafold to create plots, csv files, and chimera attribute files for each ranked model. By default output
will be saved to the same folder. See -h for more options.
[user@cn3144]$ alphapickle -od T1049
If the model above was created with the monomer_ptm model the following two plots are generated for each model:
To get an idea of runtimes of alphafold2 we first ran 4 individual proteins
on all our available GPUs. The proteins ranged in size from 144 aa to 622 aa. Note
that for all but the smallest protein, K80 GPUs were not suitable for alphafold2 and
will be deprecated from the system soon. These tests were run with default settings except for
a fixed --max_template_date=2021-07-31
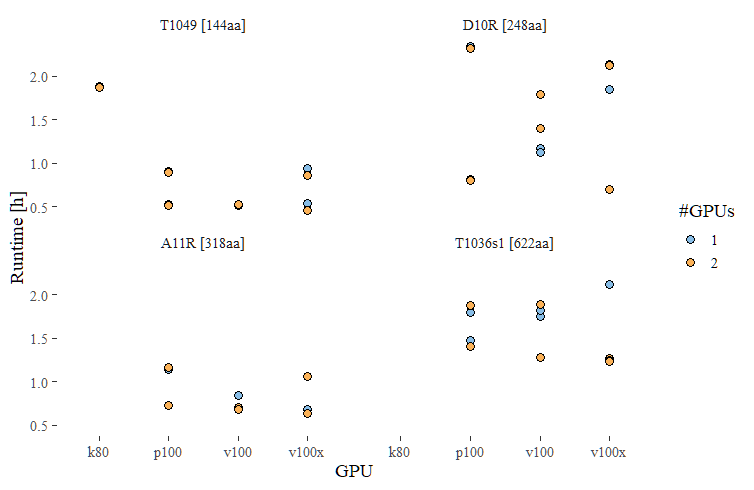
The runtime to run all 4 protein on a V100x GPU with 8 CPUs and 60GB of memory was 3.2h, slightly less than the individual runtimes of the 4 proteins run separately. For this one job we also increased the number of CPUs to 16 or the number of GPUs to 2, neither of which appeared to shorted the runtime
The resource usage profile of the combined alphafold2 pipeline in our testing thus far is suboptimal and variable. Steps probably should be segregated into individual jobs with proper resources. We hope to optimize this in the future
Script for doing only the multiple sequence alignment.
#!/bin/bash
# this is msa.sh
module load alphafold2/2.3.2
alphafold \
--model_preset=monomer \
--fasta_paths=$ALPHAFOLD2_TEST_DATA/T1049.fasta \
--max_template_date=2023-12-31 \
--msas_only \
--output_dir=$PWD
Script for doing only the model building using the sequence alignment created above.
#!/bin/bash
# this is model.sh
module load alphafold2/2.3.2
alphafold \
--model_preset=monomer \
--fasta_paths=$ALPHAFOLD2_TEST_DATA/T1049.fasta \
--max_template_date=2023-12-31 \
--use_precomputed_msas \
--output_dir=$PWD
Submit these jobs using the Slurm sbatch command.
[user@biowulf]$ jobid=$(sbatch --cpus-per-task=6 --mem=60g --gres=lscratch:100 --time=8:00:00 msa.sh) [user@biowulf]$ echo $jobid 21865850 [user@biowulf]$ sbatch --cpus-per-task=6 --partition=gpu --mem=40g --gres=gpu:v100x:1,lscratch:100 --dependency=$jobid --time=8:00:00 model.sh # --dependency is optional if MSA is ready from last step. Check details with: sbatch --help