Streamlines protein-protein interaction screens and high-throughput modelling of higher-order oligomers using AlphaFold-Multimer.
Please also check our general guidance on alphafold-related applications.
alphapulldown (see the modules page
for more information)colabfold_search from the
colabfold module.$ALPHAPULLDOWN_TEST_DATAcat bait.fa candidates.fa > for_colabfold.fasta and
bait.fa does not end with a newline the concatenated file will be formatted incorrectly.IndexError: Job index can be no larger than XX (got YY).
you tried to submit an array job with more subjobs than there were unique complexes to analyze. Check your
input lists and make sure the size of the array matches up with the number of expected tasks.In the following example we will screen a bait against a set of candidate interaction partners (i.e. pulldown mode).
Alignment generation does not require GPUs and can be done with the either the alignment pipeline from alphafold (included in this module) or colabfold (separate module). The mmseqs based colabfold method is considerably faster. Allocate an interactive session without GPUs for the alignment step.
[user@biowulf]$ sinteractive --mem=100g --cpus-per-task=8 --gres=lscratch:100 --time=1-12
salloc.exe: Pending job allocation 46116226
salloc.exe: job 46116226 queued and waiting for resources
salloc.exe: job 46116226 has been allocated resources
salloc.exe: Granted job allocation 46116226
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn3144 are ready for job
[user@cn3144]$ module load alphapulldown/0.30.7
[user@cn3144]$ cp ${ALPHAPULLDOWN_TEST_DATA:-none}/* .
[user@cn3144]$ ls -lh
-rw-r--r-- 1 user group 1.1K Sep 12 10:50 bait.fasta
-rw-r--r-- 1 user group 7 Sep 12 20:25 bait.txt
-rw-r--r-- 1 user group 145 Sep 12 20:25 candidates.txt
-rw-r--r-- 1 user group 7.4K Sep 12 10:49 candidates.fasta
[user@cn3144]$ ### alphafold based alignment pipeline - not parallelized
[user@cn3144]$ create_individual_features.py \
--fasta_paths=bait.fasta,candidates.fasta \
--save_msa_files=True \
--output_dir=pulldown_af_msas \
--use_precomputed_msas=False \
--max_template_date=2023-12-31 \
--skip_existing=True
[...much output...]
[user@cn3144]$ exit
salloc.exe: Relinquishing job allocation 46116226
[user@biowulf]$
MSA generation takes 35h in the example above. Note that in an interactive job it cannot be parallelized. To parallelize across subunits please see the section on running a Batch job . Note that not saving msa files saves space (12.7GB/15GB in this example) but precludes their re-use in the future.
[user@biowulf]$ sinteractive --mem=128g --cpus-per-task=16 --gres=lscratch:100
[user@cn3144]$ ### concatenate in a way that guarantees that the files end with a newline
[user@cn3144]$ cat bait.fasta <(echo "") candidates.fasta <(echo "") | sed -E '/^$/d' > for_colabfold.fasta
[user@cn3144]$ module load colabfold alphapulldown
[user@cn3144]$ colabfold_search \
--threads $SLURM_CPUS_PER_TASK \
for_colabfold.fasta $COLABFOLD_DB pulldown_cf_msas
[user@cn3144]$ cd pulldown_cf_msas
[user@cn3144]$ ls -lh
total 86M
-rw-r--r-- 1 user group 7.1M Sep 12 16:29 0.a3m
-rw-r--r-- 1 user group 9.2M Sep 12 16:29 10.a3m
-rw-r--r-- 1 user group 803K Sep 12 16:29 11.a3m
-rw-r--r-- 1 user group 611K Sep 12 16:29 12.a3m
[...snip...]
[user@cn3144]$ ## rename the alignments to something more useful
[user@cn3144]$ rename_colab_search_a3m.py --fasta ../for_colabfold.fasta
Renaming 18.a3m to P62945.a3m
Renaming 20.a3m to Q9NX20.a3m
Renaming 0.a3m to P78344.a3m
Renaming 1.a3m to Q14240.a3m
[...snip...]
[user@cn3144]$ ls -lh
total 86M
-rw-r--r-- 1 user group 1.8M Sep 12 16:29 O76094.a3m
-rw-r--r-- 1 user group 9.2M Sep 12 16:29 P08240.a3m
-rw-r--r-- 1 user group 803K Sep 12 16:29 P09132.a3m
-rw-r--r-- 1 user group 2.1M Sep 12 16:29 P22090.a3m
[...snip...]
[user@cn3144]$ cd ..
[user@cn3144]$ create_individual_features.py \
--fasta_paths=bait.fasta,candidates.fasta \
--output_dir=pulldown_cf_msas \
--use_precomputed_msas=True \
--max_template_date=2023-01-01 \
--use_mmseqs2=True \
--skip_existing=False
[user@cn3144]$ ## exiting the sinteractive allocation using CPUs
[user@cn3144]$ exit
salloc.exe: Relinquishing job allocation 46116226
[user@biowulf]$
The local colabfold alignment pipleine above finished in 2.5h.
Notes on the alignment step:
--use_mmseqs2=True without pre-computing alignments
with the local colabfold pileine.colabfold_search works more efficiently the more sequences are provided in a single
query. Therefore all sequences for a pulldown should be submitted as a single job.This stage of the process will require a GPU. On a A100 GPU the full example with 2 predictions per model woud take ≈ 17 h (1 bait * 20 candidates * 5 models/interaction * 2 predictions/model * 300s/prediction). Therefore we only run the first 2 candidates in the example below. The output for this step in this example is about 8.6GB/pair (172GB for all 20 pairs). If we had run only 1 prediction per model per pair it would be half that.
[user@biowulf]$ sinteractive --mem=50g -c8 --gres=lscratch:50,gpu:a100:1
[user@cn3144]$ module load alphapulldown
[user@cn3144]$ head -2 candidates.txt > 2candidates.txt
[user@cn3144]$ ## using more predictions / model will proportionally increase runtime
[user@cn3144]$ run_multimer_jobs.py \
--mode=pulldown \
--num_cycle=3 \
--num_predictions_per_model=2 \
--output_path=pulldown_models \
--protein_lists=bait.txt,2candidates.txt \
--monomer_objects_dir=pulldown_cf_msas
[user@cn3144]$ exit
salloc.exe: Relinquishing job allocation 46116226
[user@biowulf]$
Custom mode can be used to explicitly define which interactions to test and can be used to only analyze fragments of a sequence. This can be used, for example, to screen for interaction domains in proteins within very large proteins. In our example we will use one of the highly scoring pairs above and map the interaction between the full length bait and three fragments of Q14240 to illustrate the procedure. Running from the same directory as the examples above.
[user@biowulf]$ sinteractive --mem=50g -c8 --gres=lscratch:50,gpu:a100:1
[user@cn3144]$ module load alphapulldown
[user@cn3144]$ cat <<__EOF__ > custom.txt
P78344;Q14240,1-130
P78344;Q14240,131-260
P78344;Q14240,161-407
__EOF__
[user@cn3144]$ run_multimer_jobs.py \
--mode=custom \
--num_cycle=3 \
--num_predictions_per_model=2 \
--output_path=custom \
--protein_lists=custom.txt \
--monomer_objects_dir=pulldown_cf_msas
[user@cn3144]$ exit
salloc.exe: Relinquishing job allocation 46116226
[user@biowulf]$
alphapulldown has a script that will collect metrics from a run and create a jupyter notebook. Starting
this interactive session with --tunnel so that we can immediately look at the
notebook. See our Jupyter documentation
on how to connect to the notebook.
[user@biowulf]$ sinteractive --tunnel --cpus-per-task=2 --mem=10g
[user@cn3144]$ module load alphapulldown
[user@cn3144]$ create_notebook.py --help
USAGE: /opt/conda/envs/app/bin/create_notebook.py [flags]
flags:
/opt/conda/envs/app/bin/create_notebook.py:
--[no]create_notebook: Whether creating a notebook
(default: 'true')
--cutoff: cutoff value of PAE. i.e. only pae<cutoff is counted good
(default: '5.0')
(a number)
--output_dir: directory where predicted models are stored
(default: '.')
--pae_figsize: figsize of pae_plot, default is 50
(default: '50')
(an integer)
--surface_thres: surface threshold. must be integer
(default: '2')
(an integer)
[user@cn3144]$ cd pulldown_models
[user@cn3144]$ create_notebook.py
[user@cn3144]$ alphapulldown_jupyter notebook --port $PORT1 --ip localhost --no-browser
See this partially rendered example notebook.
In addition alphapulldown provides a script to create a tabular summary of putative hits:
[user@cn3144]$ run_get_good_pae.sh --help
USAGE: /app/alpha-analysis/get_good_inter_pae.py [flags]
flags:
/app/alpha-analysis/get_good_inter_pae.py:
--cutoff: cutoff value of PAE. i.e. only pae<cutoff is counted good
(default: '5.0')
(a number)
--output_dir: directory where predicted models are stored
--surface_thres: surface threshold. must be integer
(default: '2')
(an integer)
[user@cn3144]$ run_get_good_pae.sh --output_dir .
[...much output...]
[user@cn3144]$ head -2 predictions_with_good_interpae.csv
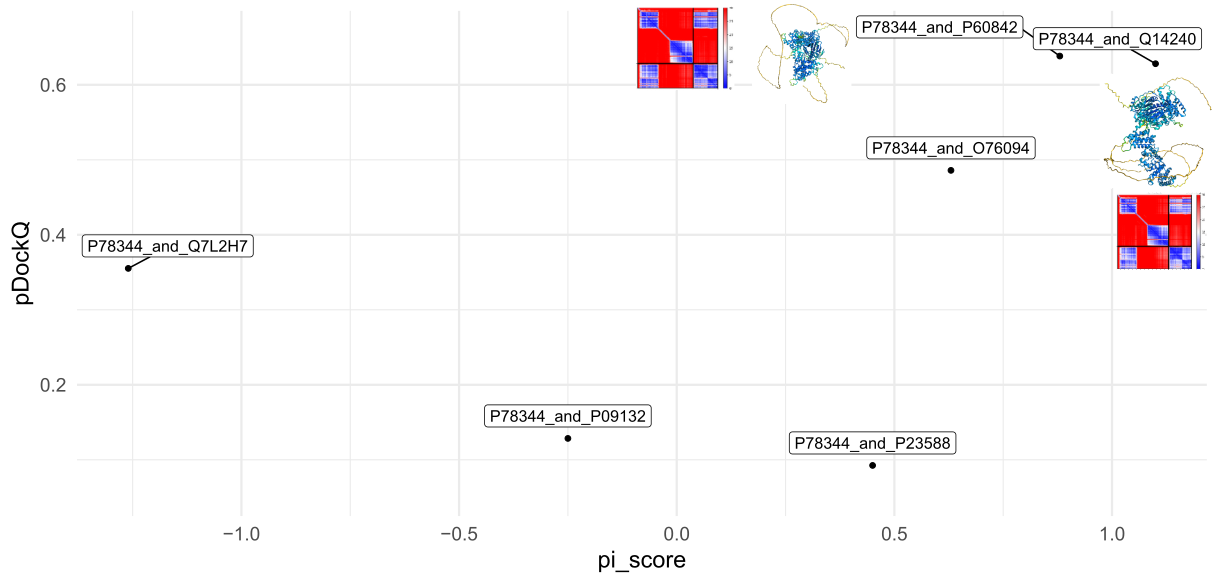
jobs,interface,Num_intf_residues,Polar,Hydrophobhic,Charged,contact_pairs, sc, hb, sb, int_solv_en, int_area,pi_score,pdb, pvalue,iptm_ptm,iptm,mpDockQ/pDockQ
P78344_and_P60842,C_B,100,0.23,0.25,0.39,104,0.52,32,27,-8.62,3795.28,0.88,,,0.7768986939967186,0.8371253,0.6383528254038463
[user@cn3144]$ exit
salloc.exe: Relinquishing job allocation 46116226
[user@biowulf]$
pDockQ and Protein Interface score (pi_score) from the output table above
The alphafold based alignment and the structural inference steps can be parallelized using slurm job arrays. The colabfold based alignment should submit all proteins in a single alignment job.
#! /bin/bash
set -e
module load colabfold alphapulldown
cp ${ALPHAPULLDOWN_TEST_DATA:-none}/* .
## concatenate in a way that makes sure there are newlines between sequences
cat bait.fasta <(echo "") candidates.fasta <(echo "") \
| sed -E '/^$/d' > for_colabfold.fasta
colabfold_search \
--threads $SLURM_CPUS_PER_TASK \
for_colabfold.fasta $COLABFOLD_DB pulldown_cf_msas
rename_colab_search_a3m.py --fasta for_colabfold.fasta pulldown_cf_msas/*.a3m
create_individual_features.py \
--fasta_paths=bait.fasta,candidates.fasta \
--output_dir=pulldown_cf_msas \
--use_precomputed_msas=True \
--max_template_date=2023-01-01 \
--use_mmseqs2=True \
--skip_existing=False
Submit this job using the Slurm sbatch command.
[user@biowulf]$ sbatch --cpus-per-task=16 --mem=128g --time=6:00:00 \
--gres=lscratch:50 alphapulldown_1a.sh
Alternatively you can use the alphafold2 based MSA generation pipeline.
#! /bin/bash
set -e
module load alphapulldown
cp ${ALPHAPULLDOWN_TEST_DATA:-none}/* .
create_individual_features.py \
--fasta_paths=bait.fasta,candidates.fasta \
--save_msa_files=True \
--output_dir=pulldown_af_msas \
--use_precomputed_msas=False \
--max_template_date=2023-01-01 \
--skip_existing=True \
--seq_index=$SLURM_ARRAY_TASK_ID
Submit this job using the Slurm sbatch command.
[user@biowulf]$ nbaits=$(grep -c ">" bait.fasta)
[user@biowulf]$ ## make sure nbaits makes sense
[user@biowulf]$ echo $nbaits
1
[user@biowulf]$ ncandidates=$(grep -c ">" candidates.fasta)
[user@biowulf]$ echo $ncandiates
20
[user@biowulf]$ ntotal=$(( $nbaits + $ncandidates ))
[user@biowulf]$ # Run a job array, 10 jobs at a time:
[user@biowulf]$ sbatch --cpus-per-task=8 --mem=100g --time=8:00:00 --gres=lscratch:100 \
--array=1-$ntotal%10 alphapulldown_1b.sh
#! /bin/bash
set -e
module load alphapulldown
run_multimer_jobs.py \
--mode=pulldown \
--num_cycle=3 \
--num_predictions_per_model=2 \
--output_path=pulldown_models \
--protein_lists=bait.txt,candidates.txt \
--monomer_objects_dir=pulldown_cf_msas \
--job_index=$SLURM_ARRAY_TASK_ID
Submit this job using the Slurm sbatch command.
[user@biowulf]$ nbaits=$(wc -l < bait.txt)
[user@biowulf]$ ## make sure nbaits makes sense ; missing final newlines or empty lines can result in incorrect counts
[user@biowulf]$ echo $nbaits
1
[user@biowulf]$ ncandidates=$(wc -l < candidates.txt)
[user@biowulf]$ ## make sure ncandidates makes sense ; missing final newlines or empty lines can result in incorrect counts
[user@biowulf]$ echo $ncandiates
20
[user@biowulf]$ ntotal=$(( $nbaits * $ncandidates ))
[user@biowulf]$ # Run a job array, 10 jobs at a time:
[user@biowulf]$ sbatch --cpus-per-task=8 --mem=50g --time=3:00:00 \
--gres=lscratch:50,gpu:a100:1 --partition=gpu --array=1-$ntotal%10 alphapulldown_2.sh