A practical introduction to GATK 4 on Biowulf (NIH HPC)
2023-03-09
Prerequisites
This tutorial requires a basic understanding of high throughput sequencing, genomics, high performance computing and bash scripting.
Software
The following tools are used in this tutorial:
- GATK 4.3.0.0
- fastp 0.20.1
- bwa 0.7.17
- samtools 1.11
- mosdepth 0.3.0
All are available on Biowulf as modules.
Sequencing data
In this tutorial we will analyze a trio from the Coriell CEPH/UTAH 1463 pedigree. The sequencing data is part of the illumina platinum genomes project (Eberle et al. 2017).
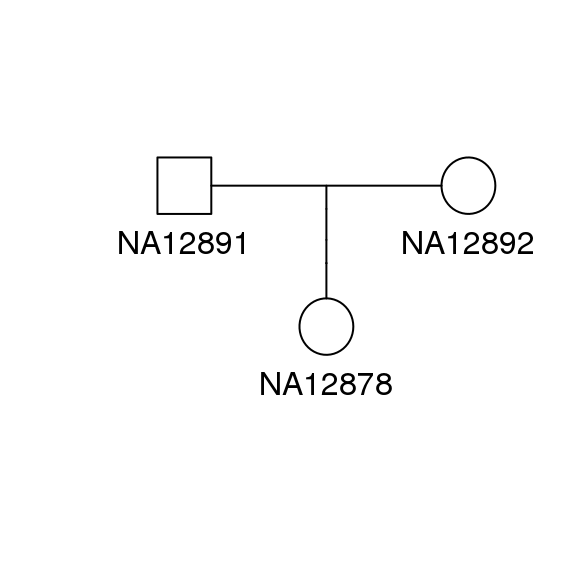
Figure 0.1: Pedigree of the family sequenced for this tutorial (CEPH pedigree 1463)
Data is available from the European Nucleotide archive under accession ERP001960 and dbGAP under accession phs001224.v1.p1.
For convenience, data for the three individuals used in this tutorial are available
on Biowulf at /fdb/app_testdata/fastq/Homo_sapiens/platinum_genomes split by
flowcell and lane to make assignment of read groups during alignment easier.
| Individual | EBI accession | Type | Pair count |
|---|---|---|---|
| NA12891 | ERR194160 | PE100 | 775,617,169 |
| NA12892 | ERR194161 | PE100 | 843,454,257 |
| NA12878 | ERR194147 | PE100 | 787,265,109 |
To run the whole pipeline, you will need about 700GB in your /data directory. Please run checkquota to make sure you have enough disk storage. If not, please request a storage increase here.