Chapter 3 MarkDuplicates
3.1 Brief introduction
Duplicate reads are derived from the same original physical fragment in the DNA library. There are two types of duplicates: PCR duplicates and Sequencing (various optical confusions) duplicates. To avoid measurement bias and reduce error, only one best copy should be kept.
To take only one representative read, GATK uses a Picard tool
(MarkDuplicates) to mark all the other reads from a set of duplicates with
a tag. Reads are tagged but not removed from the alignment.
Here we use MarkDuplicatesSpark instead of MarkDuplicates. Spark is
used for parallelism in GATK 4 and can speed up the process relative to the
serial tools. However, not all Spark based tools are considered production
quality and there aren’t Spark versions of each GATK tool. To use Spark
multithreading on the Biowulf cluster, it is necessary to add --spark-master local[$SLURM_CPUS_ON_NODE] to the base command line.
MarkDuplicatesSpark is optimized to run on queryname-grouped alignments
(that is, all reads with the same queryname are together in the input file). If
provided coordinate-sorted alignments (output of samtools sort or picard SortSam), the tool will spend additional time first queryname sorting the reads
internally.
3.2 Benchmarks of MarkDuplicatesSpark
3.2.1 Queryname-grouped input data (as generated by the aligner)
We did a benchmark on the performance of MarkDuplicatesSpark on queryname-grouped input with different numbers of CPUs and memory. As show in figure 3.1, the runtime is not reduced much when using more than 20 threads.These benchmark tests are performed on the same type of nodes (x2680).
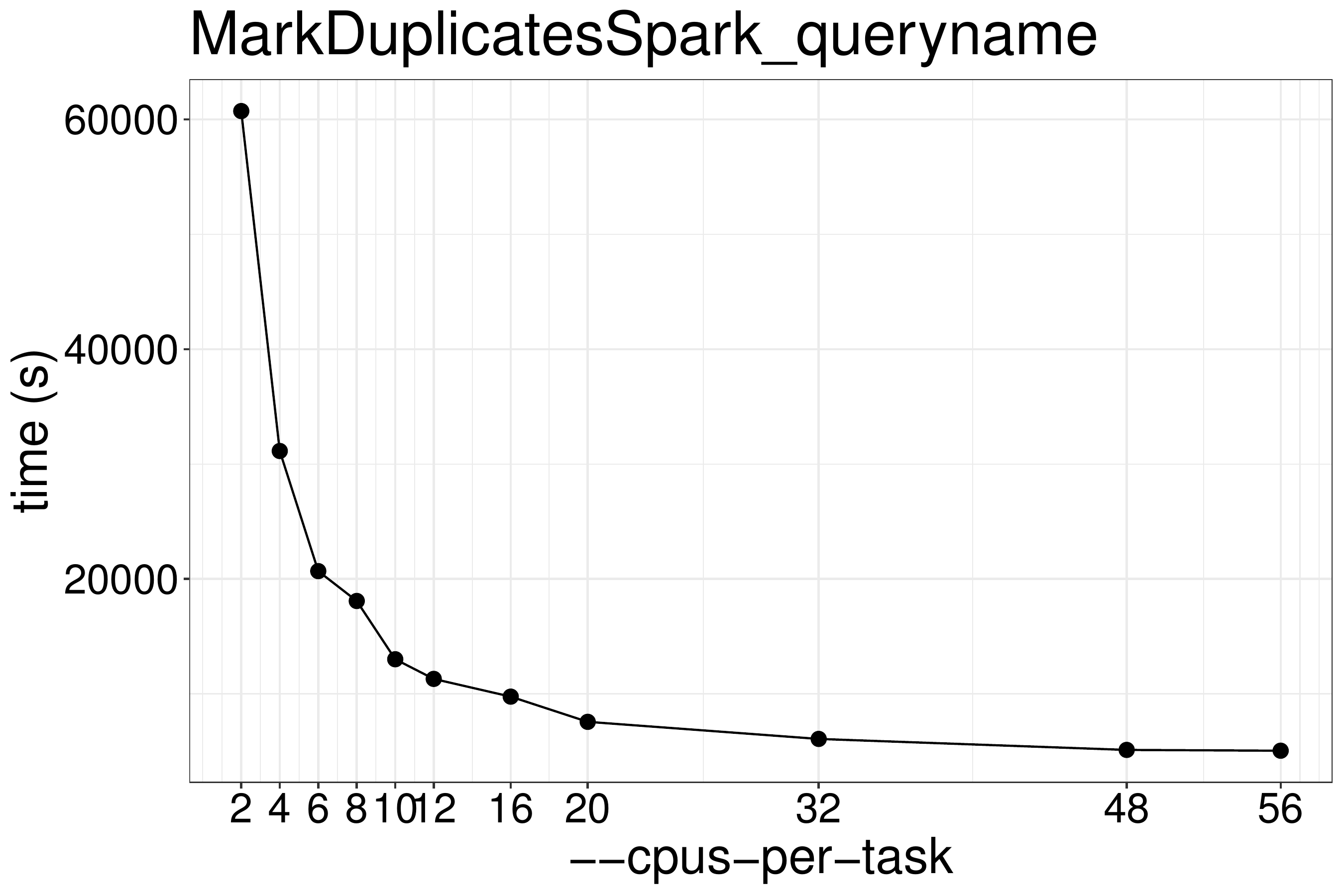
Figure 3.1: MarkDuplicatesSpark runtime with different numbers of threads.
We normally recommend to run scripts with 70%-80% effiency (figure
3.2), therefore MarkDuplicatesSpark should be
run with no more than 20 threads. Using the G1GC garbage collector did not improve performance with default parameters.
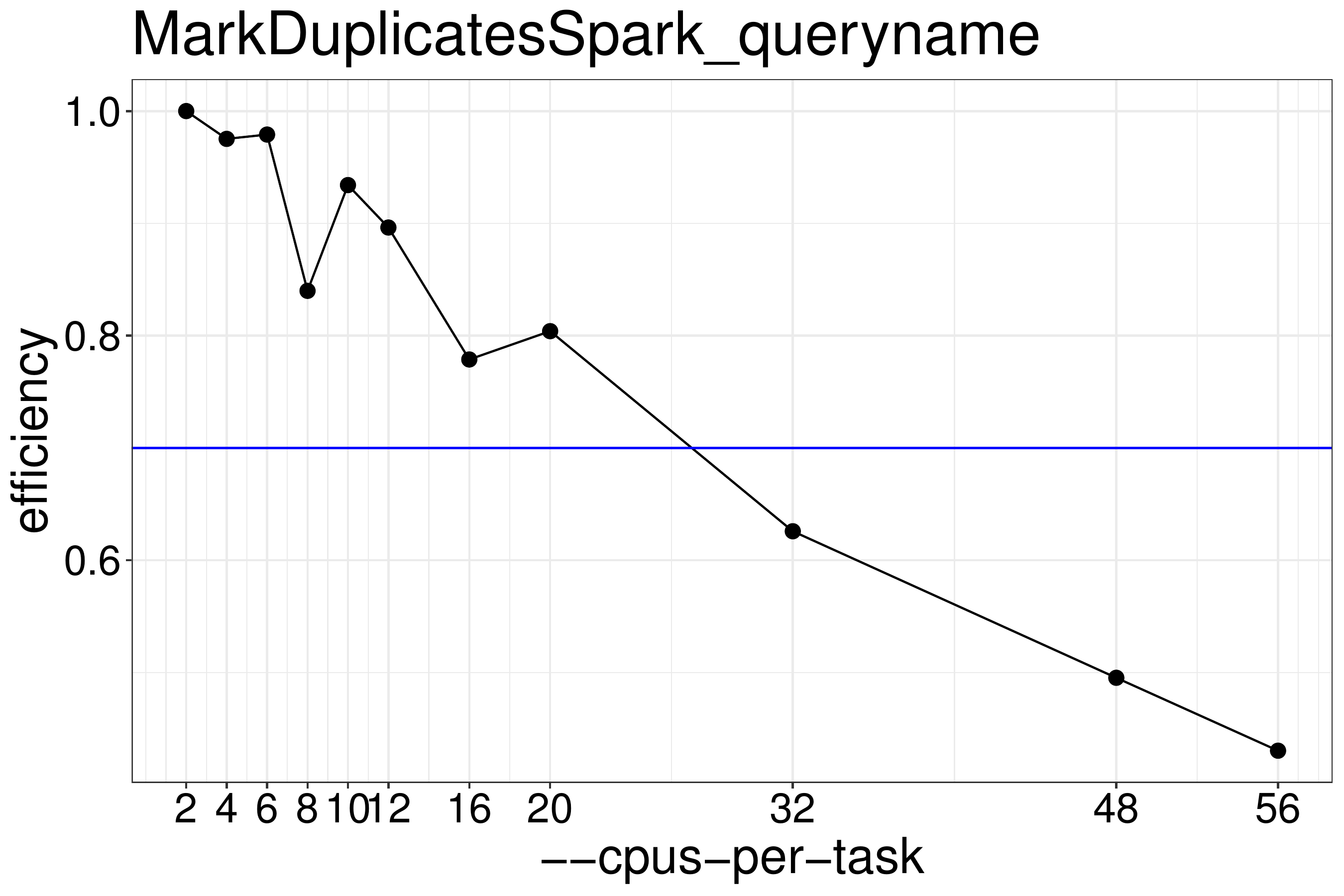
Figure 3.2: MarkDuplicatesSpark efficiency for queryname-grouped input data. For good efficiency no more than 20 threads should be run.
As for memory, increasing memory beyond the required minimum of 80GB (20 threads) did not improve the performance (figure 3.3).
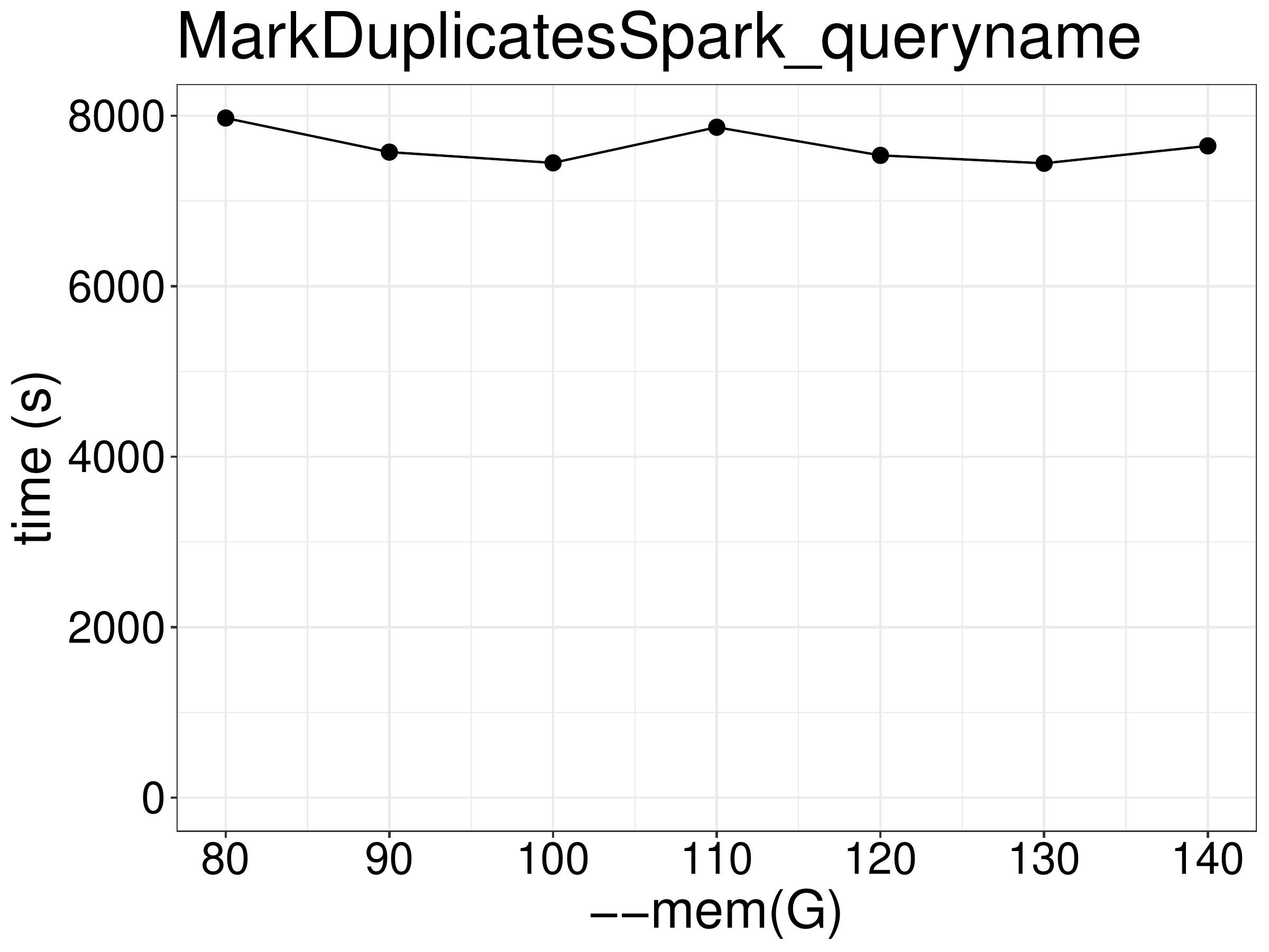
Figure 3.3: Increasing memory did not improve performance.
3.2.2 Coordinate-sorted input data
We also did a benchmark on the performance of MarkDuplicatesSpark on
coordinate-sorted input files with different numbers of CPUs and memory. As
show in figure 3.4, the runtime is not reduced much
when using more than 8 threads. These benchmark tests are performed on the same
type of nodes (x2680).
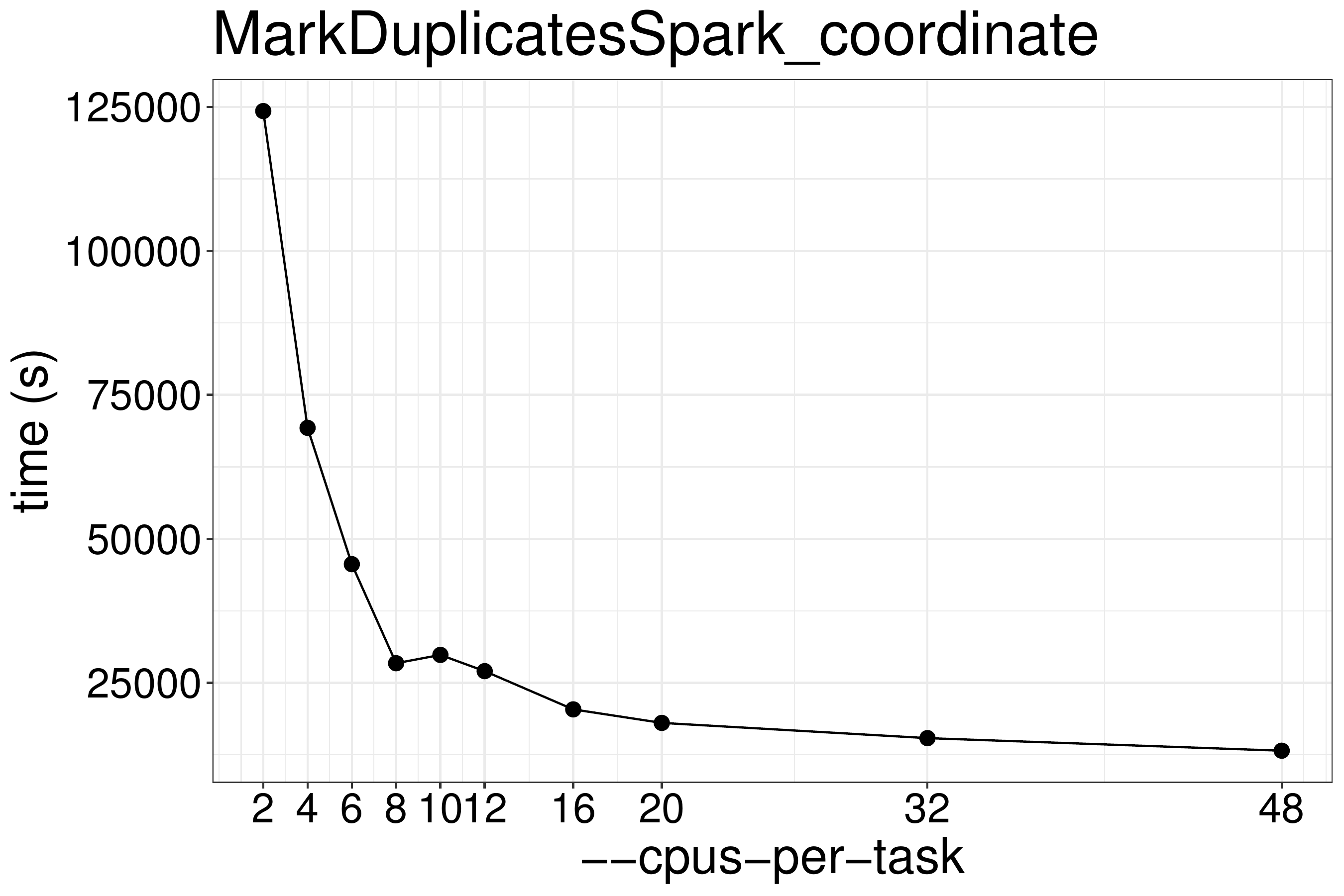
Figure 3.4: MarkDuplicatesSpark runtime with different numbers of threads.
We normally recommend to run scripts with 70%-80% efficiency (figure 3.5), therefore MarkDuplicatesSpark should be run with no more than 16 threads.
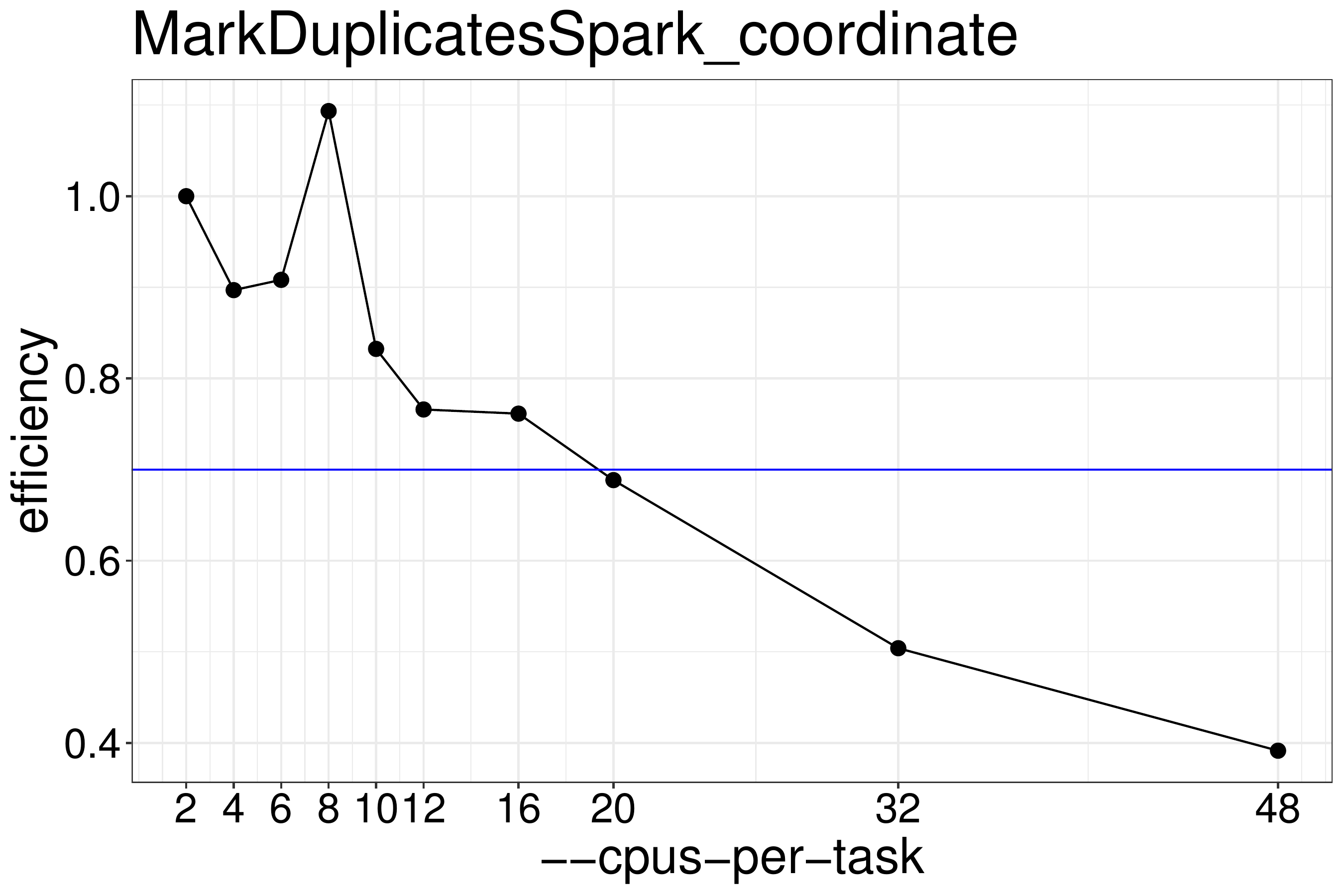
Figure 3.5: MarkDuplicatesSpark efficiency for coordinate-sorted input data. For good efficiency no more than 16 threads should be run.
Increasing memory to more than the required minimum of 60GB (at 8 threads) did not improve performance (figure 3.6).
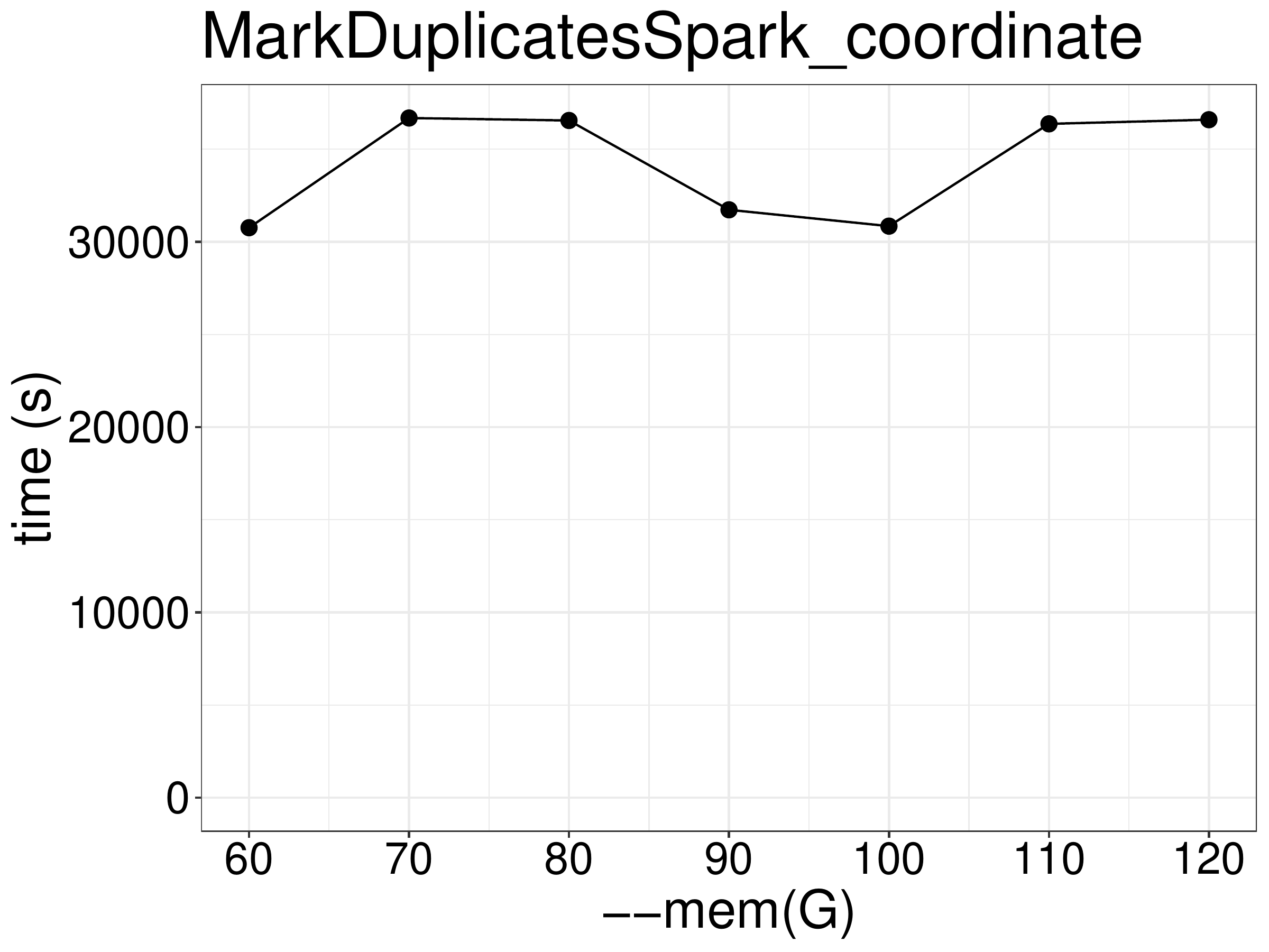
Figure 3.6: Increasing memory did not improve performance.
3.2.3 Performance comparing between queryname-grouped and coordinate-sorted inputs
Our test shows that using coordinate-sorted inputs can increase processing time as much as 2-fold (figure 3.7).
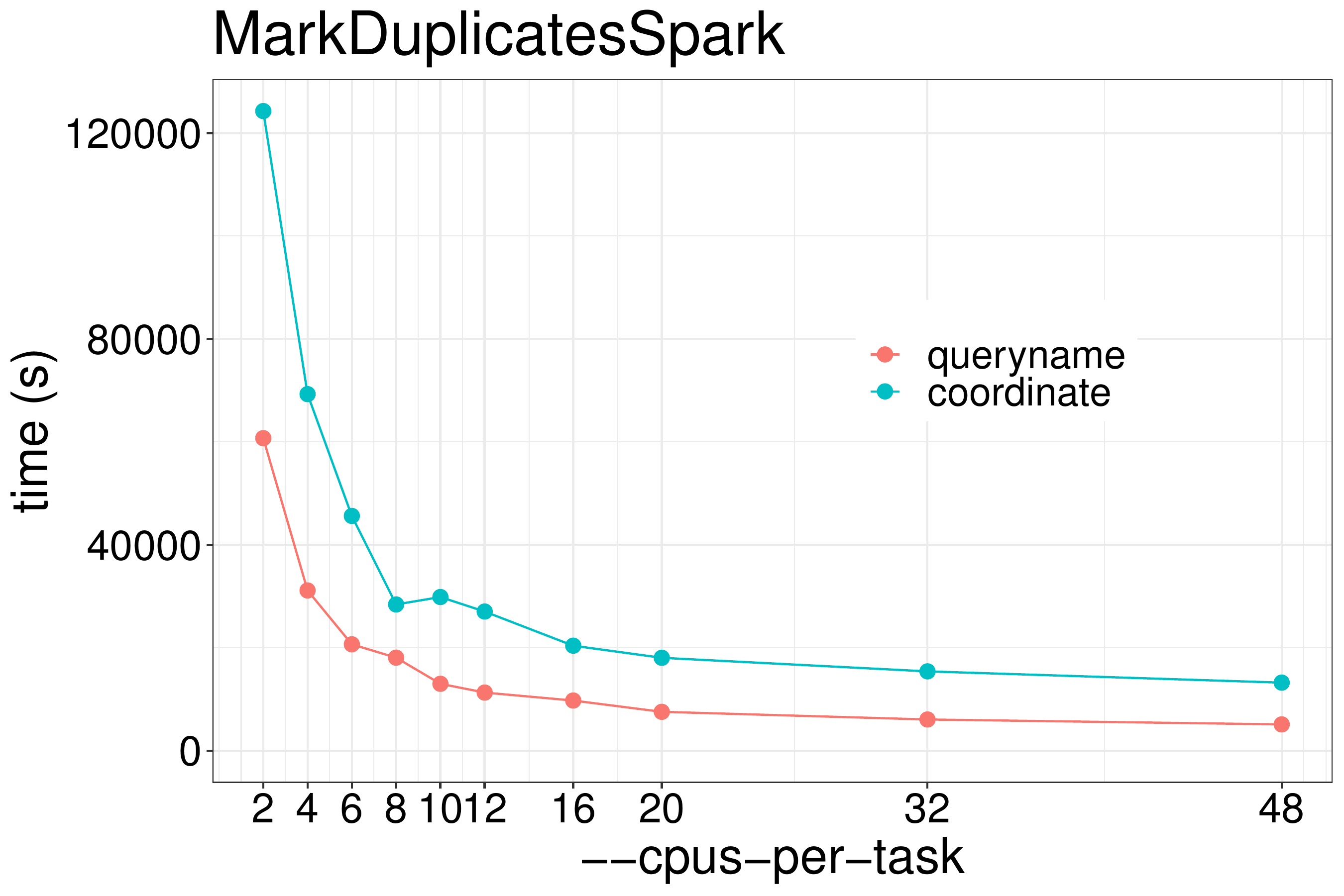
Figure 3.7: Using queryname grouped input unmodified from the aligner vs coordinate sorted input is as much as 2x faster.
3.3 Optimized script
Your first real GATK commands:
# run MarkDuplicatesSpark as swarm jobs
cd your_input_dir/ || exit ; \
cp SRR622457_bwa.bam /lscratch/$SLURM_JOBID/; \
if [[ $SLURM_CPUS_ON_NODE -gt 12 ]]; \
then gatk --java-options "-Djava.io.tmpdir=/lscratch/$SLURM_JOBID -Xms60G -Xmx60G" MarkDuplicatesSpark \
-I /lscratch/$SLURM_JOBID/SRR622457_bwa.bam \
-O /lscratch/$SLURM_JOBID/SRR622457_bwa_markdup.bam \
--spark-master local[12]; \
else gatk --java-options "-Djava.io.tmpdir=/lscratch/$SLURM_JOBID -Xms60G -Xmx60G" MarkDuplicatesSpark \
-I /lscratch/$SLURM_JOBID/SRR622457_bwa.bam \
-O /lscratch/$SLURM_JOBID/SRR622457_bwa_markdup.bam \
--spark-master local[$SLURM_CPUS_ON_NODE]; \
fi; \
cp /lscratch/$SLURM_JOBID/SRR622457_bwa_markdup.bam your_output_dir/
cd your_input_dir/ || exit ; \
cp SRR622458_bwa.bam /lscratch/$SLURM_JOBID/; \
if [[ $SLURM_CPUS_ON_NODE -gt 12 ]]; \
then gatk --java-options "-Djava.io.tmpdir=/lscratch/$SLURM_JOBID -Xms60G -Xmx60G" MarkDuplicatesSpark \
-I /lscratch/$SLURM_JOBID/SRR622458_bwa.bam \
-O /lscratch/$SLURM_JOBID/SRR622458_bwa_markdup.bam \
--spark-master local[12]; \
else gatk --java-options "-Djava.io.tmpdir=/lscratch/$SLURM_JOBID -Xms60G -Xmx60G" MarkDuplicatesSpark \
-I /lscratch/$SLURM_JOBID/SRR622458_bwa.bam \
-O /lscratch/$SLURM_JOBID/SRR622458_bwa_markdup.bam \
--spark-master local[$SLURM_CPUS_ON_NODE]; \
fi; \
cp /lscratch/$SLURM_JOBID/SRR622458_bwa_markdup.bam your_output_dir/
cd your_input_dir/ || exit ; \
cp SRR622458_bwa.bam /lscratch/$SLURM_JOBID/; \
if [[ $SLURM_CPUS_ON_NODE -gt 12 ]]; \
then gatk --java-options "-Djava.io.tmpdir=/lscratch/$SLURM_JOBID -Xms60G -Xmx60G" MarkDuplicatesSpark \
-I /lscratch/$SLURM_JOBID/SRR622459_bwa.bam \
-O /lscratch/$SLURM_JOBID/SRR622459_bwa_markdup.bam \
--spark-master local[12]; \
else gatk --java-options "-Djava.io.tmpdir=/lscratch/$SLURM_JOBID -Xms60G -Xmx60G" MarkDuplicatesSpark \
-I /lscratch/$SLURM_JOBID/SRR622459_bwa.bam \
-O /lscratch/$SLURM_JOBID/SRR622459_bwa_markdup.bam \
--spark-master local[$SLURM_CPUS_ON_NODE]; \
fi; \
cp /lscratch/$SLURM_JOBID/SRR622459_bwa_markdup.bam your_output_dir/You can submit these commands as separate Biowulf jobs with swarm, but please do not exclusively allocating nodes due the decline of efficiency on large number of CPUs:
swarm -t 14 -g 80 --gres=lscratch:800 --time=12:00:00 -m GATK/4.3.0.0 -f 03-GATK_MarkDuplicatesSpark.shNotes:
- GATK will print a lot of console output. The header will tell you what you’re running; the progress meter will inform you how far along you are; and the footer will signal when the run is complete and running time.
- If anything went wrong, check the output files first.
- Common errors include typos and incorrect file paths.