Chapter 2 GATK practice workflow
Here we build a workflow for germline short variant calling. It is based on the GATK Best Practices workshop taught by the Broad Institute which was also the source of the figures used in this Chapter.
There are three main steps: Cleaning up raw alignments, joint calling, and variant filtering.
2.1 Cleaning up raw alignments
This includes two steps:
- Step 1: Marking duplicate reads (
MarkDuplicates,MarkDuplicatesSpark) (Chapter 3) -
Marking duplicates is a general preprocessing step for variant calling. Most variant
detection tools require duplicates to be tagged in mapped reads to reduce bias. - Step 2: Base Quality Scores Recalibration (
BaseRecalibrator,ApplyBQSR) (Chapter 4) - Sequencers make systematic errors in assigning base quality scores. To correct for these errors, a model is built using covariates encoded in the read groups from all base calls and then applying the adjustments to generate recalibrated base qualities(2.1).
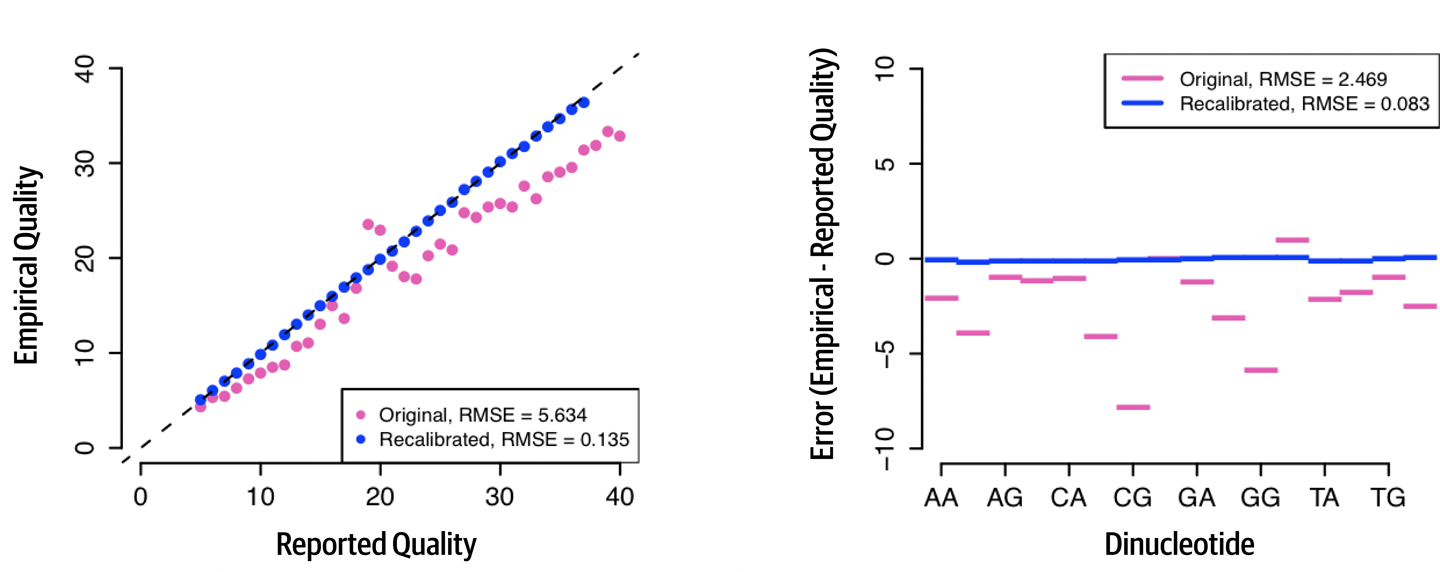
Figure 2.1: The effect of BQSR
2.2 Joint Calling
A single genome is rarely useful by itself. Most scientific questions require the analysis of multiple genomes - anywhere from a few genomes from a family to study disease inheritance to large numbers in population genetic studies. The joint calling approach takes advantage of data from the whole set of genomes to improve sensitivity of genotype inference in each genome, boost statistical power and reduce technical artifacts. It accounts, for example, for the difference between no variant call in a sample due to missing data vs good data showing no variation.
It also makes it easier to add samples progressively by combining initial calls in GVCF or GenomicsDB format.
This example uses a parent-daughter trio. Single sample analysis is not discussed.
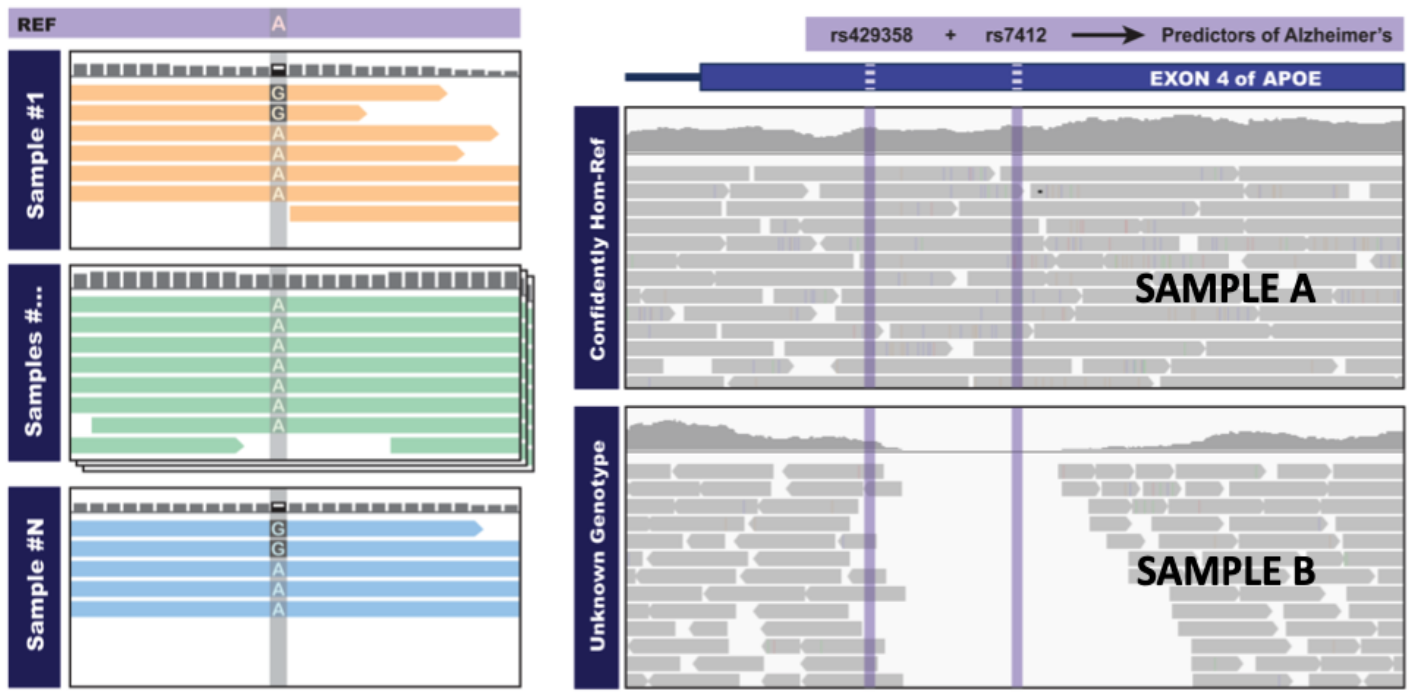
Figure 2.2: The power of joint calling
Example (2.2): (left) The variant allele is present in only two of the N samples, in both cases with low coverage so that the variant may not be callable when processed separately. (right) Neither sample will have records in a variants-only output file when analyzed separately, they will be identified as non-informative. The first sample is homozygous reference, while the second sample has no data. In both cases, joint calling allows evidence to be accumulated over all samples.
There are three steps in joint callings:
2.3 Variant filtering
Raw variant calls include many artifacts and the goal of variant filtering is to remove as many artifacts as possible at the minimal loss of sensitivity for real variants.
- Step 1: Variant Quality Score Recalibration (
VariantRecalibrator,ApplyVQSR) (Chapter 8) - The core algorithm in VQSR is a Gaussian mixture model that aims to classify variants based on how their annotation values cluster given a training set of high-confidence variants(2.3).
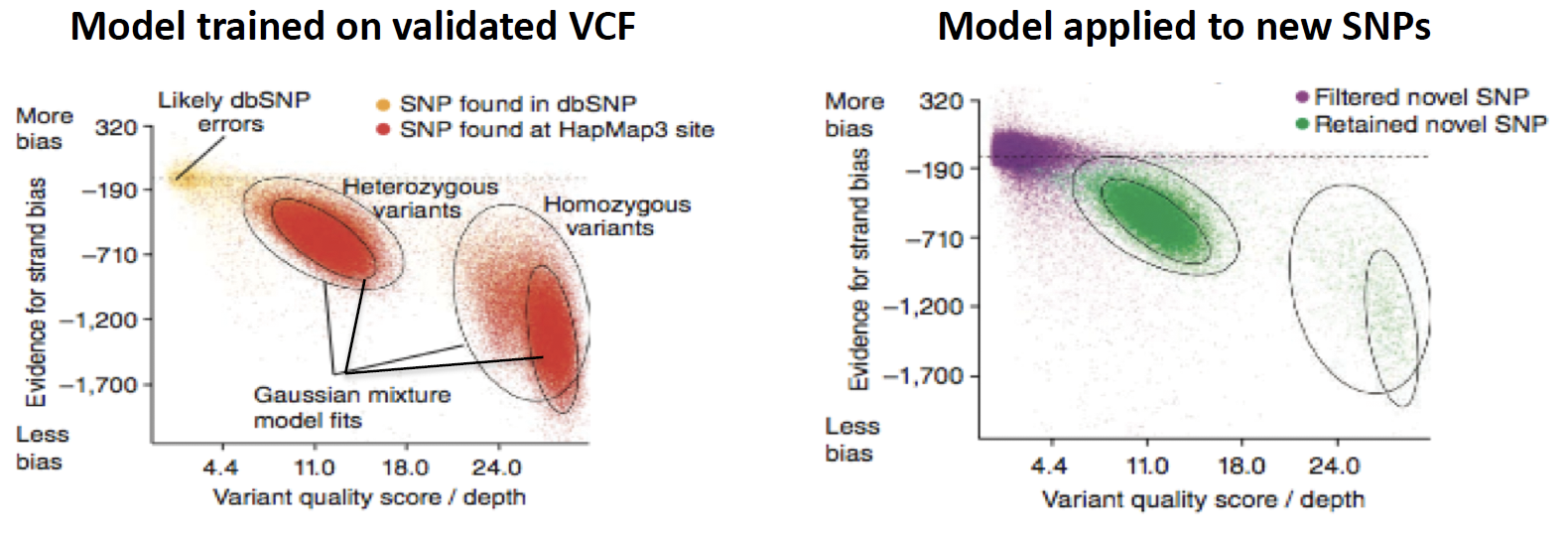
Figure 2.3: Determination of VQSL shreshold
- Step 2:
CalculateGenotypePosteriors(Chapter 9) - Uses pedigree information and allele frequencies to refine genotype calls.