Chapter 8 VQSR
8.1 Brief introduction
Raw variant calls include many artifacts.
The core algorithm in VQSR is a Gaussian mixture model that aims to classify variants based on how their annotation values cluster given a training set of high-confidence variants.
Then the VQSR tools use this model to assign a new confidence score to each variant, called VQSLOD. This is a log-ratio of the variant’s probabilities belonging to the positive and negative model.
8.2 Benchmarks of VariantRecalibrator
We did a benchmark on the performance of VariantRecalibrator with different
numbers of CPUs and memory. As show in figure 8.1, the running
time didn’t reduce much after increasing threads.
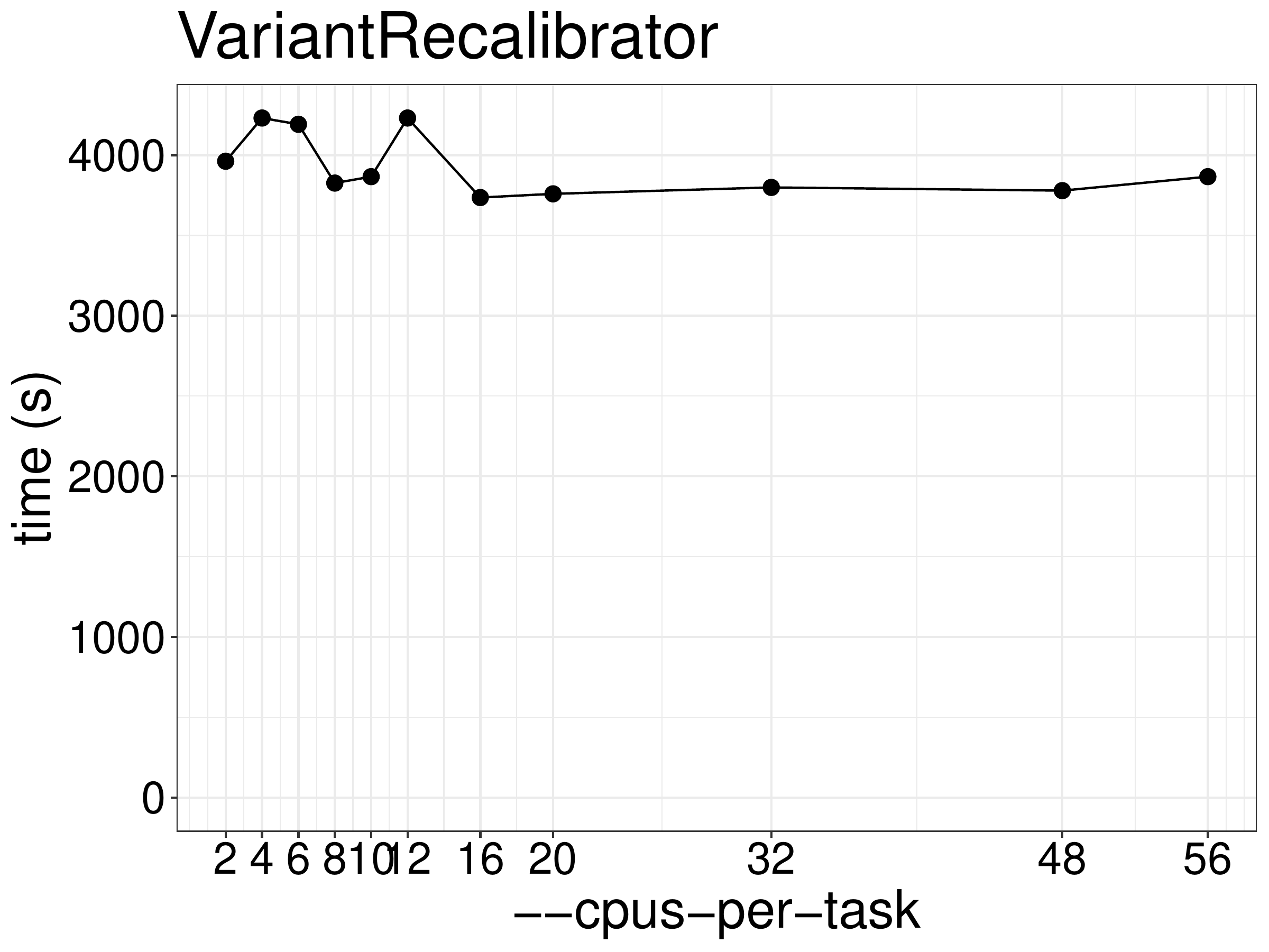
Figure 8.1: Runtime of VariantRecalibrator as a function of the number of threads
We normally recommend running jobs with 70%-80% efficiency. Based on the efficiency calculated from the runtime (figure 8.2),
variantRecalibrator should not be run with more than 2 threads.
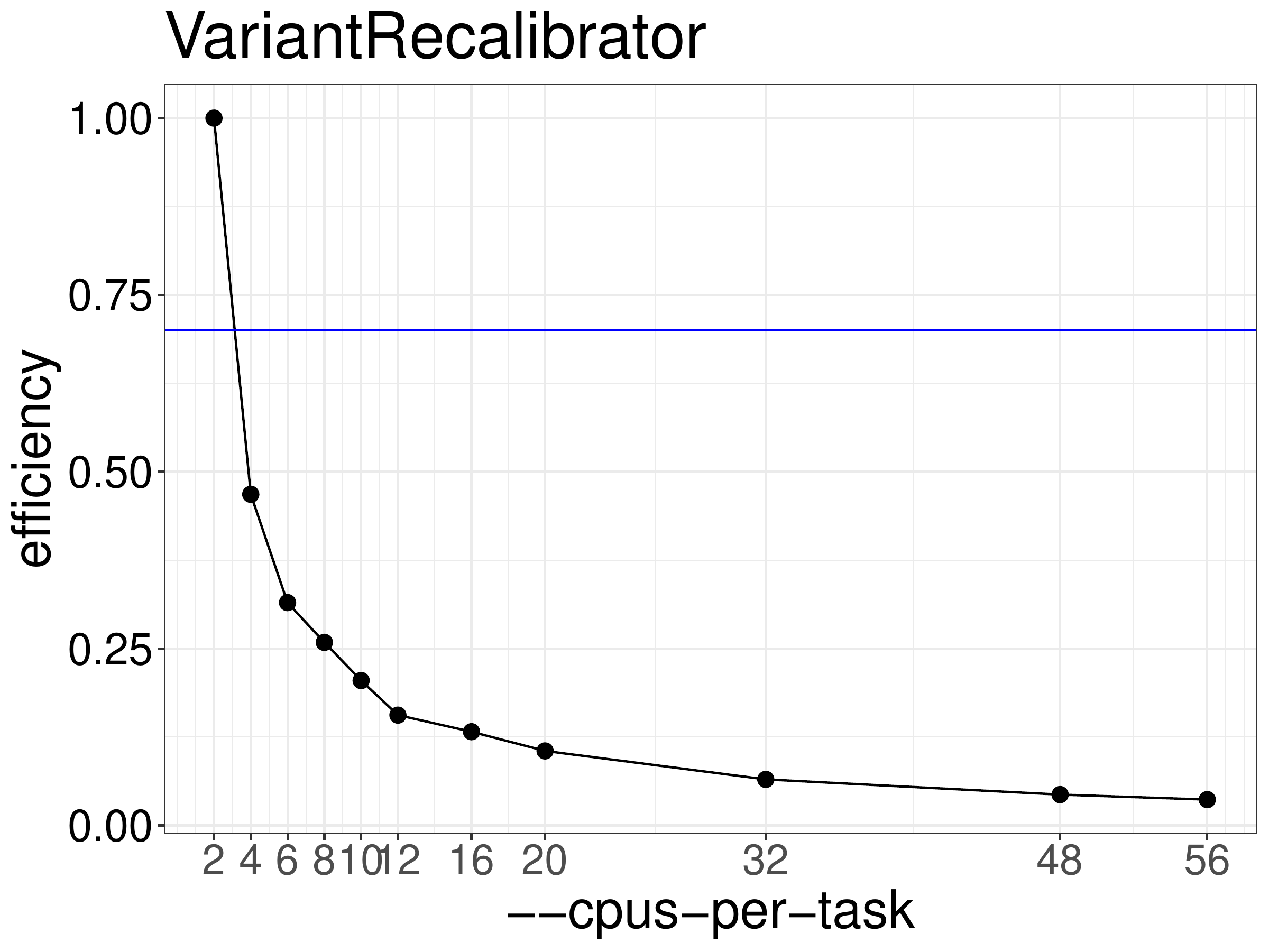
Figure 8.2: Efficiency of VariantRecalibrator as a function of the number of threads
As for memory, increasing memory didn’t improve performance (figure 8.3), although a minimal of 4GB memory is required for the job to finish with 2 threads.
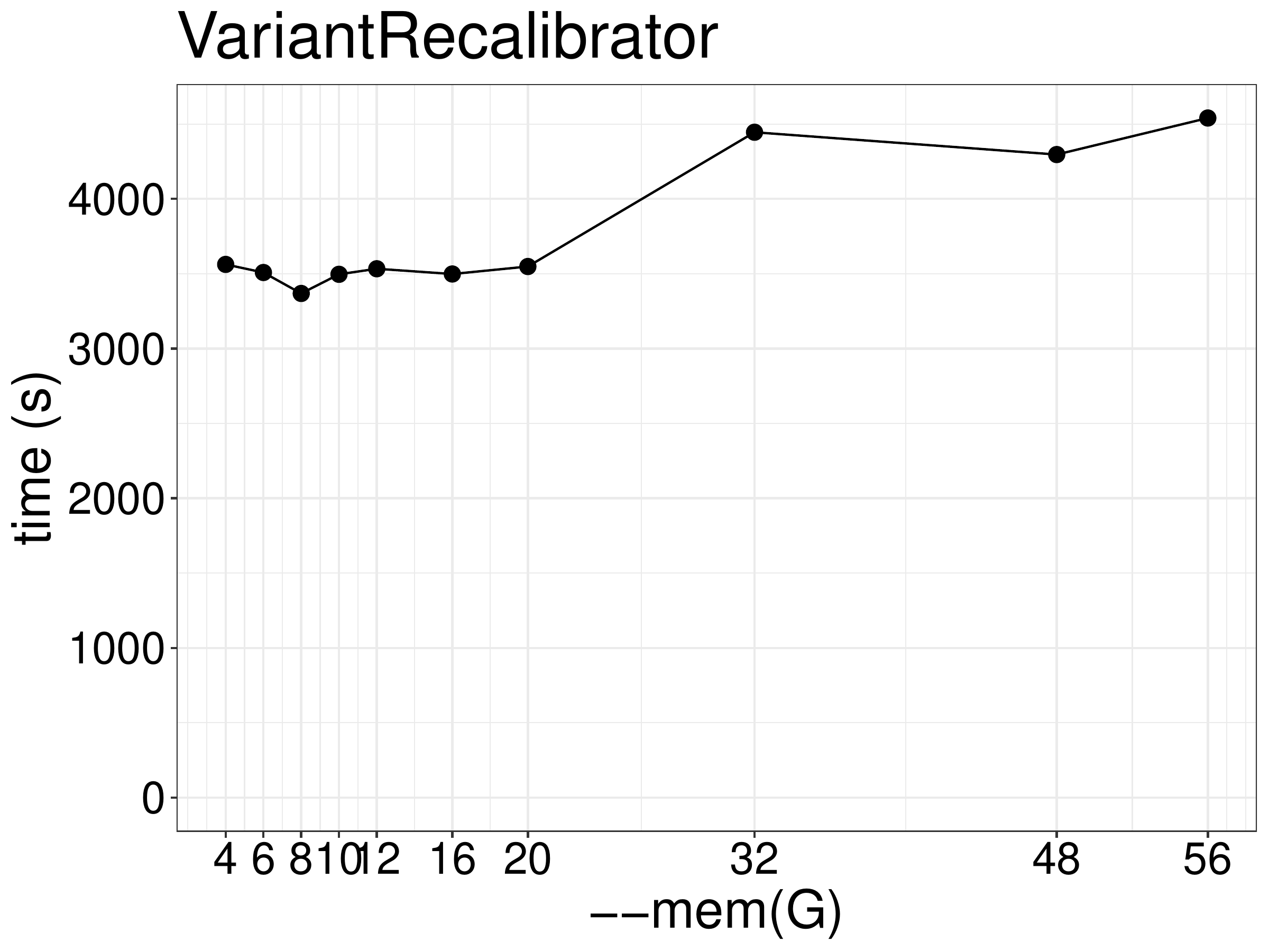
Figure 8.3: Runtime of VariantRecalibrator as a function of memory
8.3 Optimized script for VariantRecalibrator
Step 1: We use VariantRecalibrator to build the model and calculate VQSLOD
for SNP and INDEL separately:
cd data/; \
gatk --java-options "-Djava.io.tmpdir=/lscratch/$SLURM_JOBID -Xms4G -Xmx4G -XX:ParallelGCThreads=2" VariantRecalibrator \
-tranche 100.0 -tranche 99.95 -tranche 99.9 \
-tranche 99.5 -tranche 99.0 -tranche 97.0 -tranche 96.0 \
-tranche 95.0 -tranche 94.0 \
-tranche 93.5 -tranche 93.0 -tranche 92.0 -tranche 91.0 -tranche 90.0 \
-R /fdb/igenomes/Homo_sapiens/UCSC/hg38/Sequence/WholeGenomeFasta/genome.fa \
-V merged.vcf.gz \
--resource:hapmap,known=false,training=true,truth=true,prior=15.0 \
/fdb/GATK_resource_bundle/hg38/hapmap_3.3.hg38.vcf.gz \
--resource:omni,known=false,training=true,truth=false,prior=12.0 \
/fdb/GATK_resource_bundle/hg38/1000G_omni2.5.hg38.vcf.gz \
--resource:1000G,known=false,training=true,truth=false,prior=10.0 \
/fdb/GATK_resource_bundle/hg38/1000G_phase1.snps.high_confidence.hg38.vcf.gz \
-an QD -an MQ -an MQRankSum -an ReadPosRankSum -an FS -an SOR \
-mode SNP -O merged_SNP1.recal --tranches-file output_SNP1.tranches \
--rscript-file output_SNP1.plots.R
cd data/; \
gatk --java-options "-Djava.io.tmpdir=/lscratch/$SLURM_JOBID -Xms4G -Xmx4G -XX:ParallelGCThreads=2" VariantRecalibrator \
-tranche 100.0 -tranche 99.95 -tranche 99.9 \
-tranche 99.5 -tranche 99.0 -tranche 97.0 -tranche 96.0 \
-tranche 95.0 -tranche 94.0 -tranche 93.5 -tranche 93.0 \
-tranche 92.0 -tranche 91.0 -tranche 90.0 \
-R /fdb/igenomes/Homo_sapiens/UCSC/hg38/Sequence/WholeGenomeFasta/genome.fa \
-V merged.vcf.gz \
--resource:mills,known=false,training=true,truth=true,prior=12.0 \
/fdb/GATK_resource_bundle/hg38/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz \
--resource:dbsnp,known=true,training=false,truth=false,prior=2.0 \
/fdb/GATK_resource_bundle/hg38/dbsnp_146.hg38.vcf.gz \
-an QD -an MQRankSum -an ReadPosRankSum -an FS -an SOR -an DP \
-mode INDEL -O merged_indel1.recal --tranches-file output_indel1.tranches \
--rscript-file output_indel1.plots.RJobs can be submitted with swarm:
swarm -t 2 -g 4 --gres=lscratch:400 --time=2:00:00 -m GATK/4.3.0.0 -f 08-GATK_VariantRecalibrator.sh8.4 Benchmarks of ApplyVQSR
We did benchmarks on the performance of ApplyVQSR with different numbers of
CPUs and memory. As show in figure 8.4, the running time is
not reduced much when using multiple threads as with the other essentially single
threaded tools.
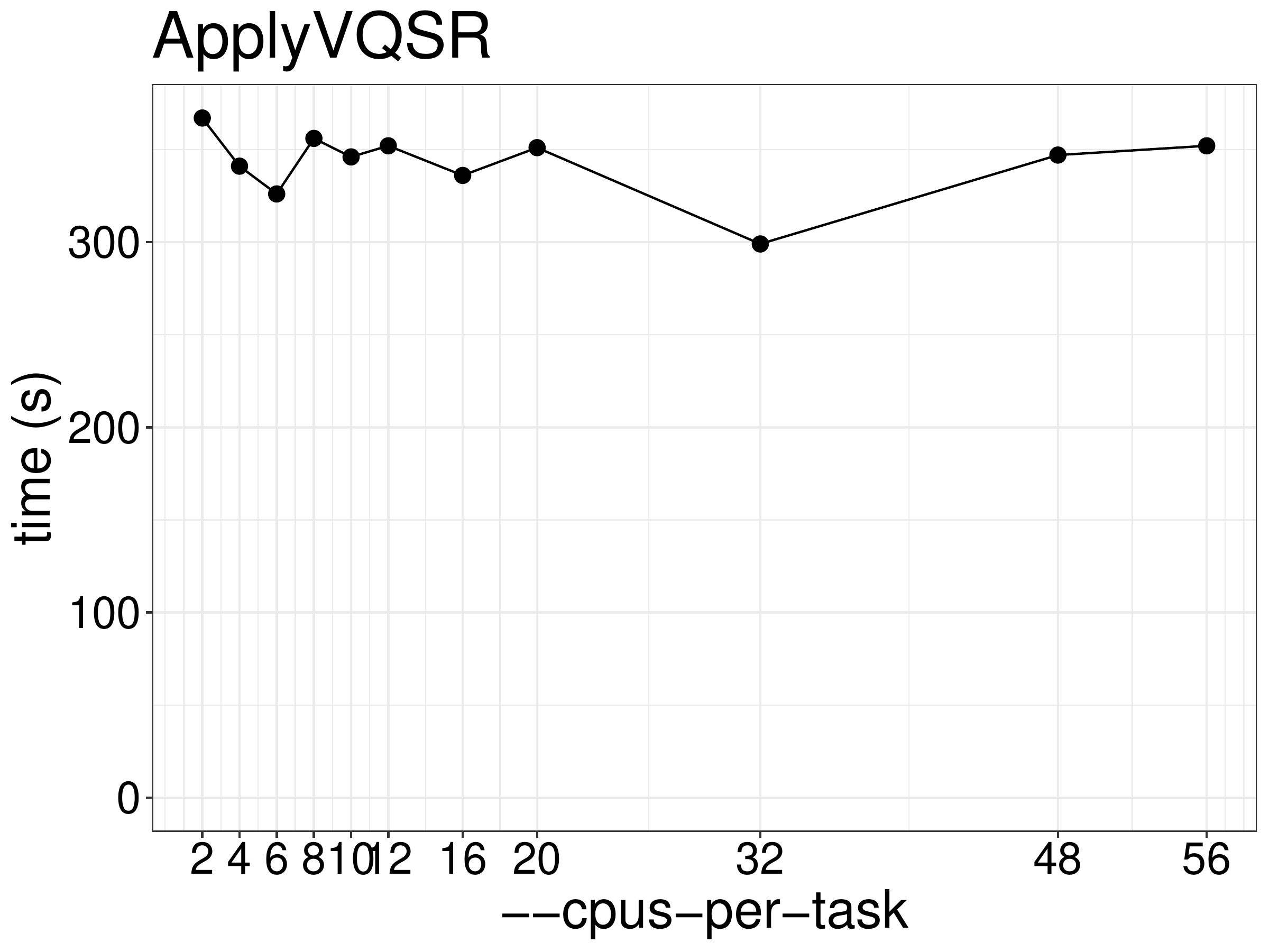
Figure 8.4: Runtime of ApplyVQSR as a function of the number of threads
We normally recommend running jobs with 70%-80% efficiency. Based on figure 8.5 we recommend running ApplyVQSR with
no more than two threads.
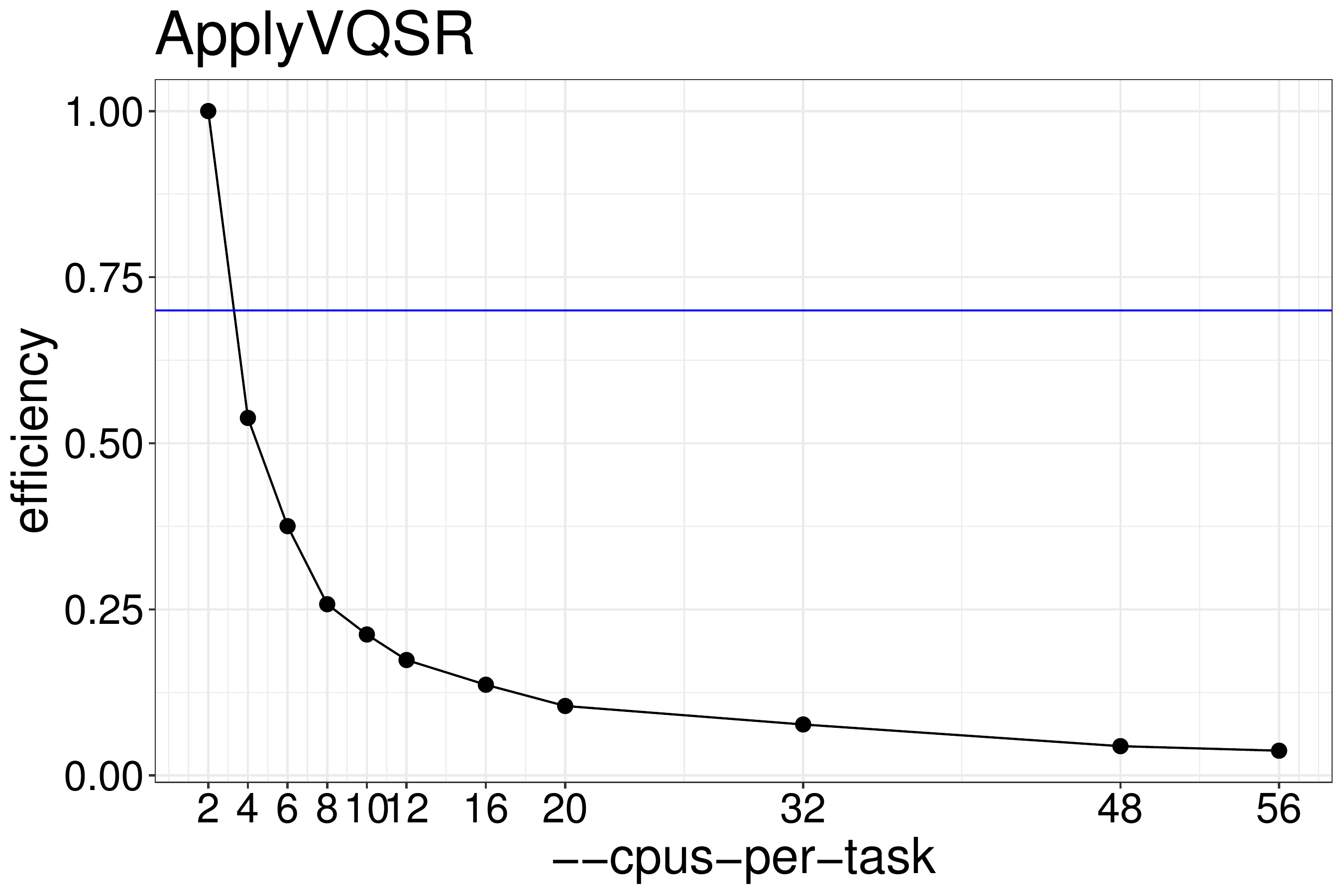
Figure 8.5: Efficiency of ApplyVQSR as a function of the number of threads
Increasing memory beyond the minimum needed did not improve performance
(figure 8.6). Using the G1GC garbage collector did not improve performance with default parameters, however
it requires extra 6GB memory to avoid out of memory kill.
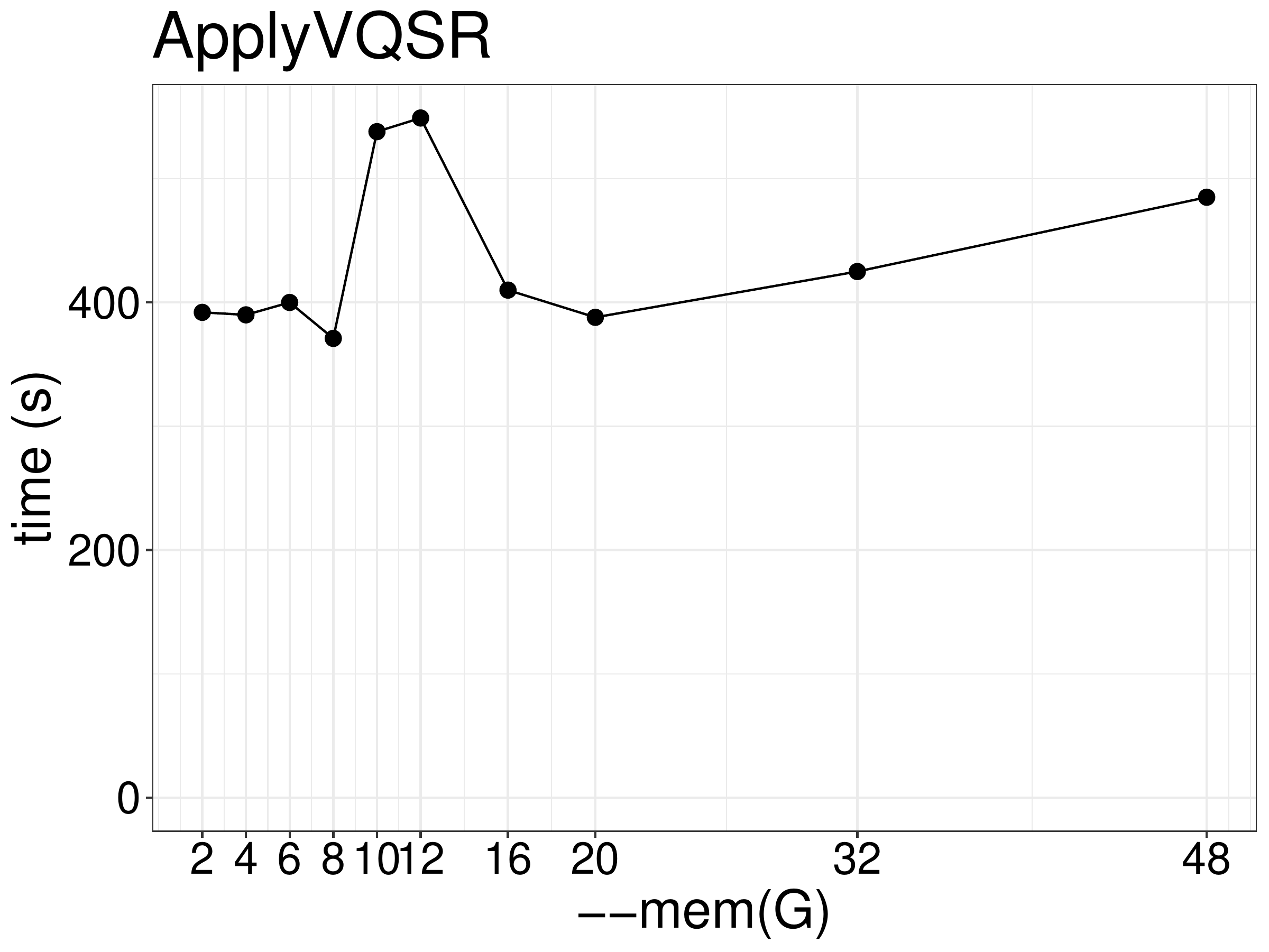
Figure 8.6: Runtime of ApplyVQSR as a function of memory
8.5 Optimized script for ApplyVQSR
Step 2: We choose the VQSLOD cutoff to filter VCF file using ApplyVQSR:
#! /bin/bash
set -euo
# run ApplyVQSR on SNP then INDEL
module load GATK/4.3.0.0
cd data/;
gatk --java-options "-Djava.io.tmpdir=/lscratch/$SLURM_JOBID \
-Xms2G -Xmx2G -XX:ParallelGCThreads=2" ApplyVQSR \
-V merged.vcf.gz \
--recal-file merged_SNP1.recal \
-mode SNP \
--tranches-file output_SNP1.tranches \
--truth-sensitivity-filter-level 99.9 \
--create-output-variant-index true \
-O SNP.recalibrated_99.9.vcf.gz
gatk --java-options "-Djava.io.tmpdir=/lscratch/$SLURM_JOBID \
-Xms2G -Xmx2G -XX:ParallelGCThreads=2" ApplyVQSR \
-V SNP.recalibrated_99.9.vcf.gz \
-mode INDEL \
--recal-file merged_indel1.recal \
--tranches-file output_indel1.tranches \
--truth-sensitivity-filter-level 99.9 \
--create-output-variant-index true \
-O indel.SNP.recalibrated_99.9.vcf.gz99.9% is the recommended default VQSLOD cutoff for SNPs in human genomic analysis.
Job submission:
sbatch --cpus-per-task=2 --mem=2g --gres=lscratch:100 --time=2:00:00 08-GATK_ApplyVQSR.shNote:
- For INDEL there is no recommended VQSLOD cutoff, here we use 99.9 just as an example.