Chapter 6 GenomicsDBImport (replaces CombineGVCFs)
6.1 Brief introduction
GVCFs are consolidated into a GenomicsDB datastore in order to improve
scalability and speedup the next step: joint genotyping. To speedup,
GenomicsDBImport was performed on each chromosome.
6.2 Benchmarks
We did a benchmark on the performance of GenomicsDBImport with different
numbers of CPUs and memory. As show in figure 6.1, the runtime
did not reduce much given more threads.
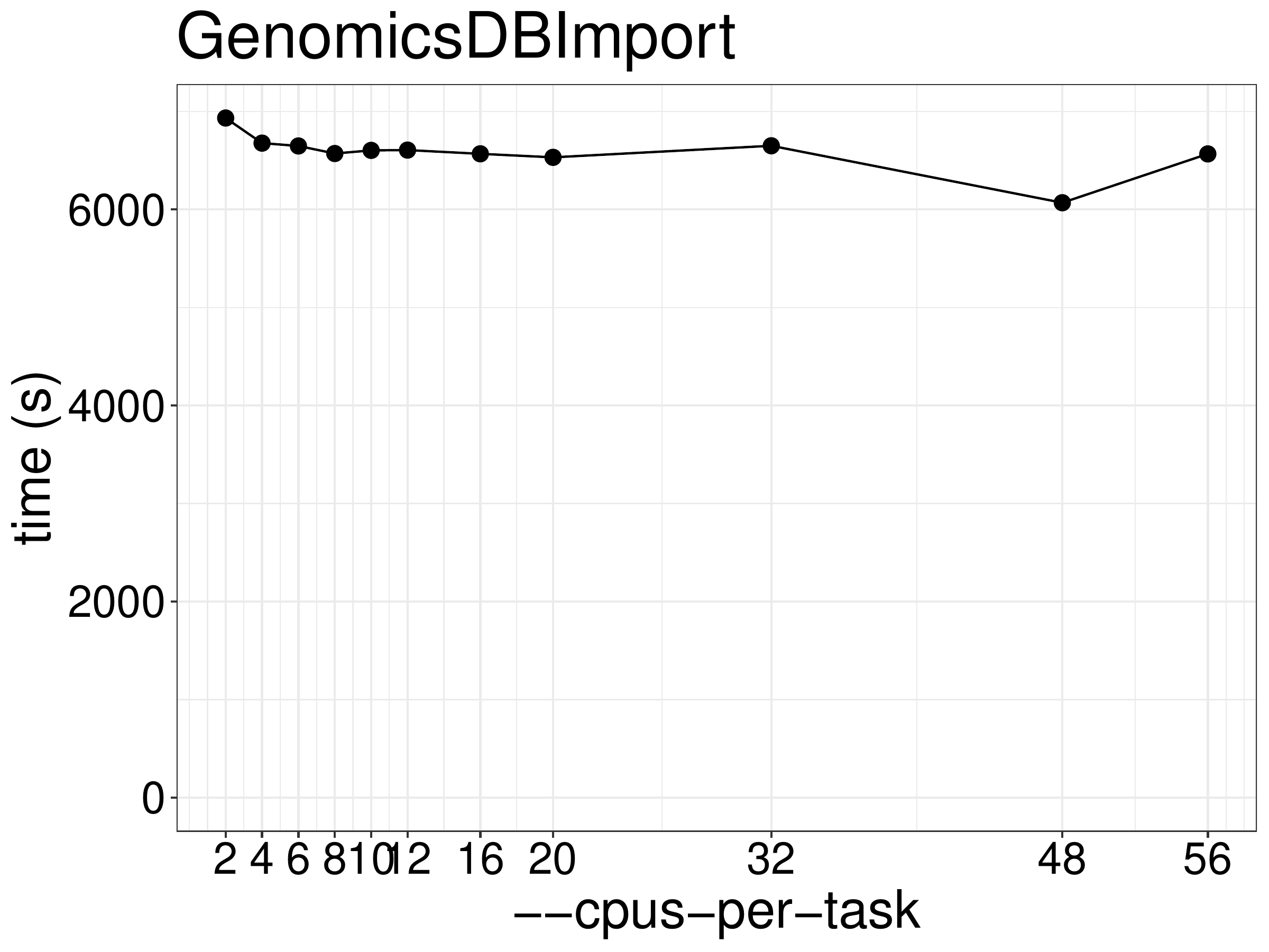
Figure 6.1: Runtime of GenomicsDBImport as a function of the number of threads
We normally recommend running jobs with 70%-80% efficiency. Based on
the efficiency calculated from the runtime results (figure 6.2) GenomicsDBImport should be run with no more than 2 threads.
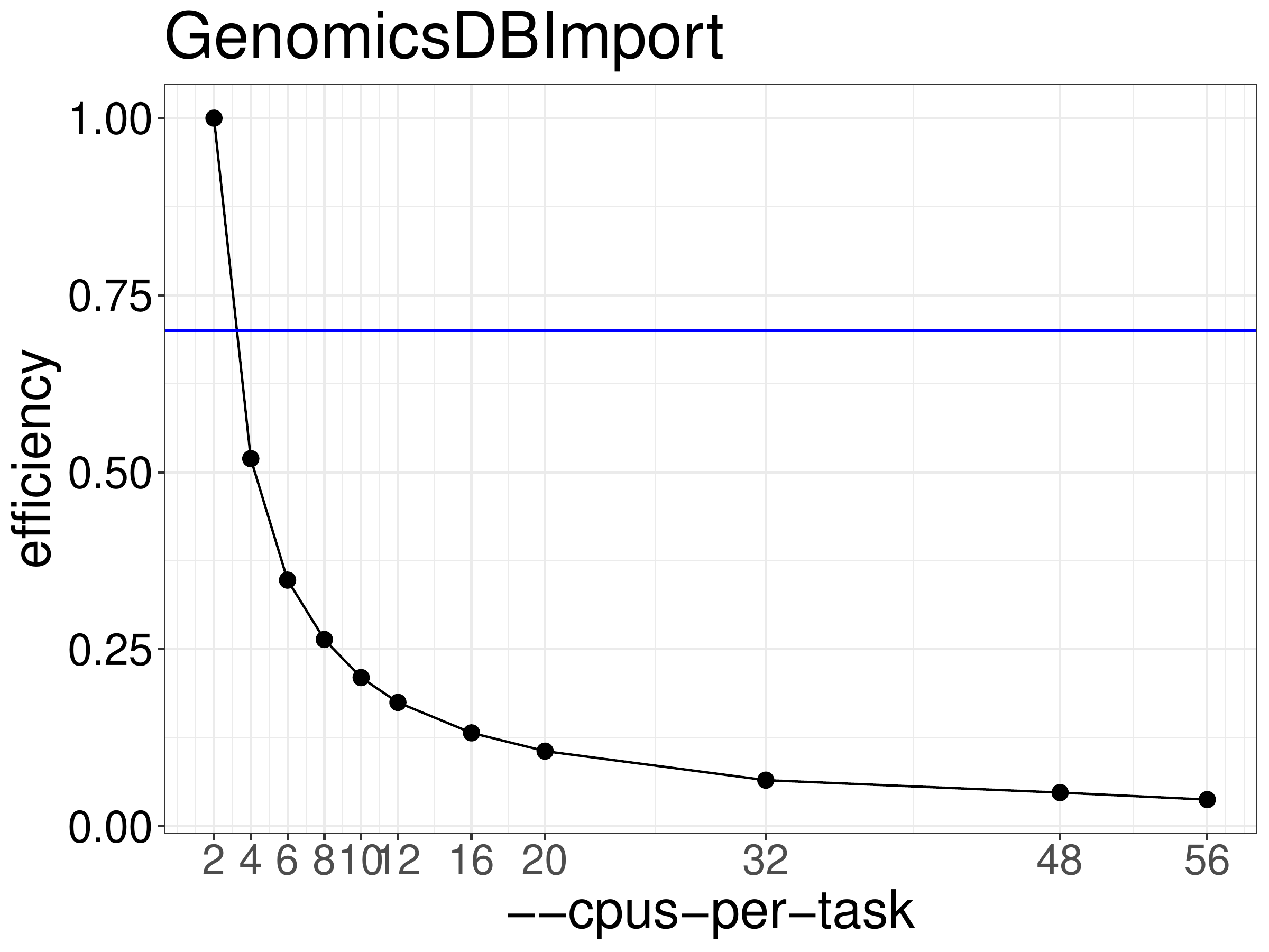
Figure 6.2: Efficiency of GenomicsDBImport as a function of the number of threads
As for memory, increasing memory didn’t improve performance (figure 6.3).
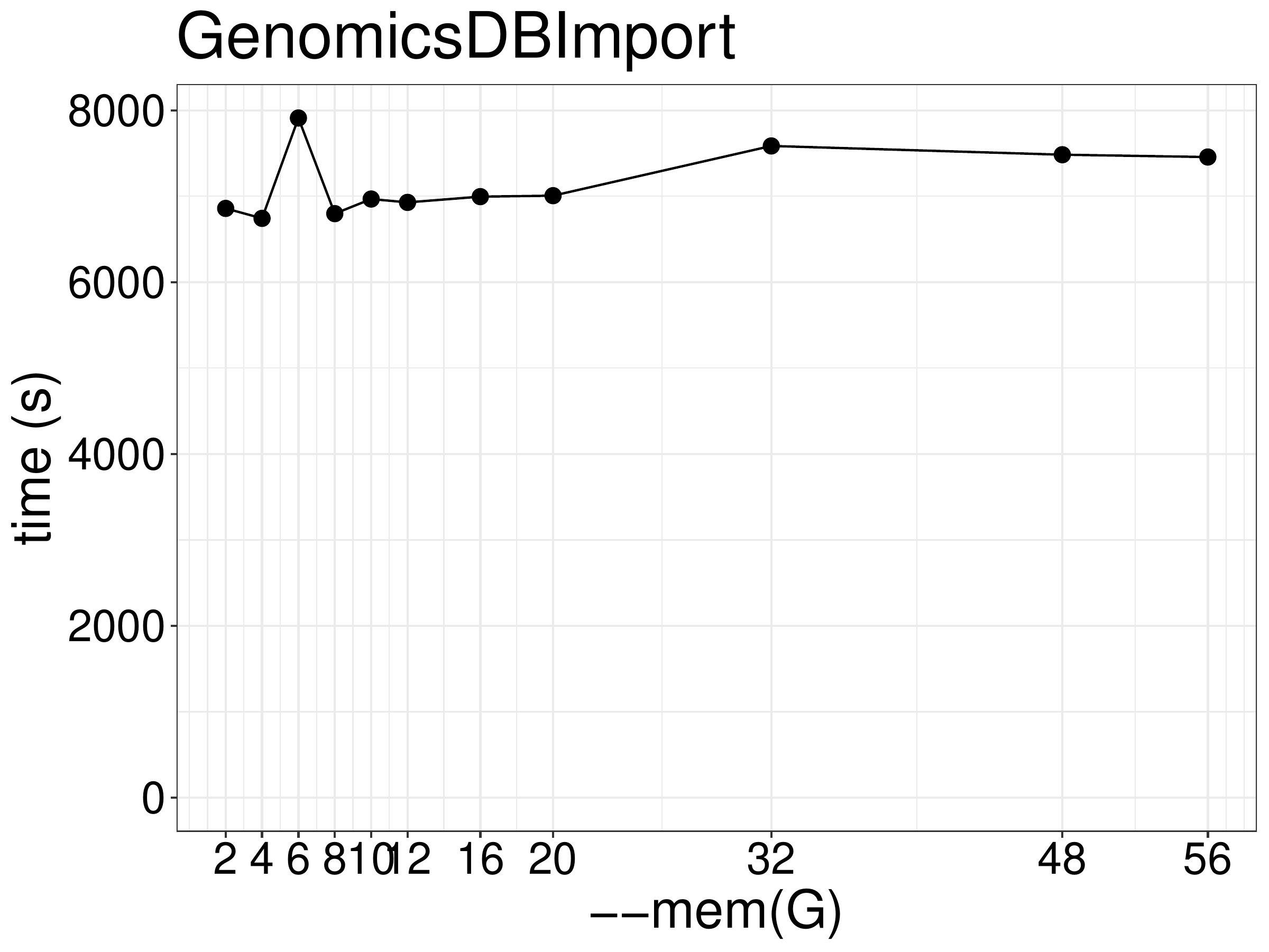
Figure 6.3: Runtime of GenomicsDBImport as a function of allocated memory.
6.3 Optimized script
#! /bin/bash
sed -e
module load GATK/4.3.0.0
for j in {1..22} X Y M; do cd data/; \
mkdir -p /lscratch/$SLURM_JOBID/tmp; \
gatk --java-options "-Djava.io.tmpdir=/lscratch/$SLURM_JOBID -Xms2G -Xmx2G -XX:ParallelGCThreads=2" GenomicsDBImport \
--genomicsdb-workspace-path /lscratch/${SLURM_JOB_ID}/chr${j}_gdb \
-R /fdb/igenomes/Homo_sapiens/UCSC/hg38/Sequence/WholeGenomeFasta/genome.fa \
-V NA12878.g.vcf.gz \
-V NA12891.g.vcf.gz \
-V NA12892.g.vcf.gz \
--tmp-dir "/lscratch/${SLURM_JOB_ID}/tmp" \
--max-num-intervals-to-import-in-parallel 3 \
--intervals chr${j}; \
# copy the final results to your data directory
cp -r /lscratch/${SLURM_JOB_ID}/chr${j}_gdb /data/$USER/data_test/gatk_ped/; doneJob submission with swarm:
sbatch --cpus-per-task=2 --mem=2g --gres=lscratch:100 --time=4:00:00 06-GATK_GenomicsDBImport.shIt is easy to add new sample to GenomicsDB, but it’s always a good idea to have a backup first.
Note:
- The GenomicsDB is difficult to examine directly, so you can use
SelectVariantsto convert it to GVCF file. - It’s important to remember that
lscratchwill be cleaned up after completing jobs, so if there is any error during the job running, there might not be any results or truncated results. Always check the status of jobs before the next step. - GenomicsDB can create very large numbers of files when created over too many intervals with many genomes.