Chapter 7 GenotypeGVCFs
7.1 Brief introduction
GenotypeGVCFs uses the potential variants from the HaplotypeCaller and does
the joint genotyping. It will look at the available information for each site
from both variant and non-variant alleles across all samples, and will produce
a VCF file containing only the sites that it found to be variant in at least
one sample.
7.2 Benchmarks
We did a benchmark on the performance of GenotypeGVCFs with different numbers
of CPUs and memory. As show in figure 7.1, the running time
did not reduce much given more threads. This is essentially a single threaded tool.
Overall speed is reduced by processing different regions in parallel.
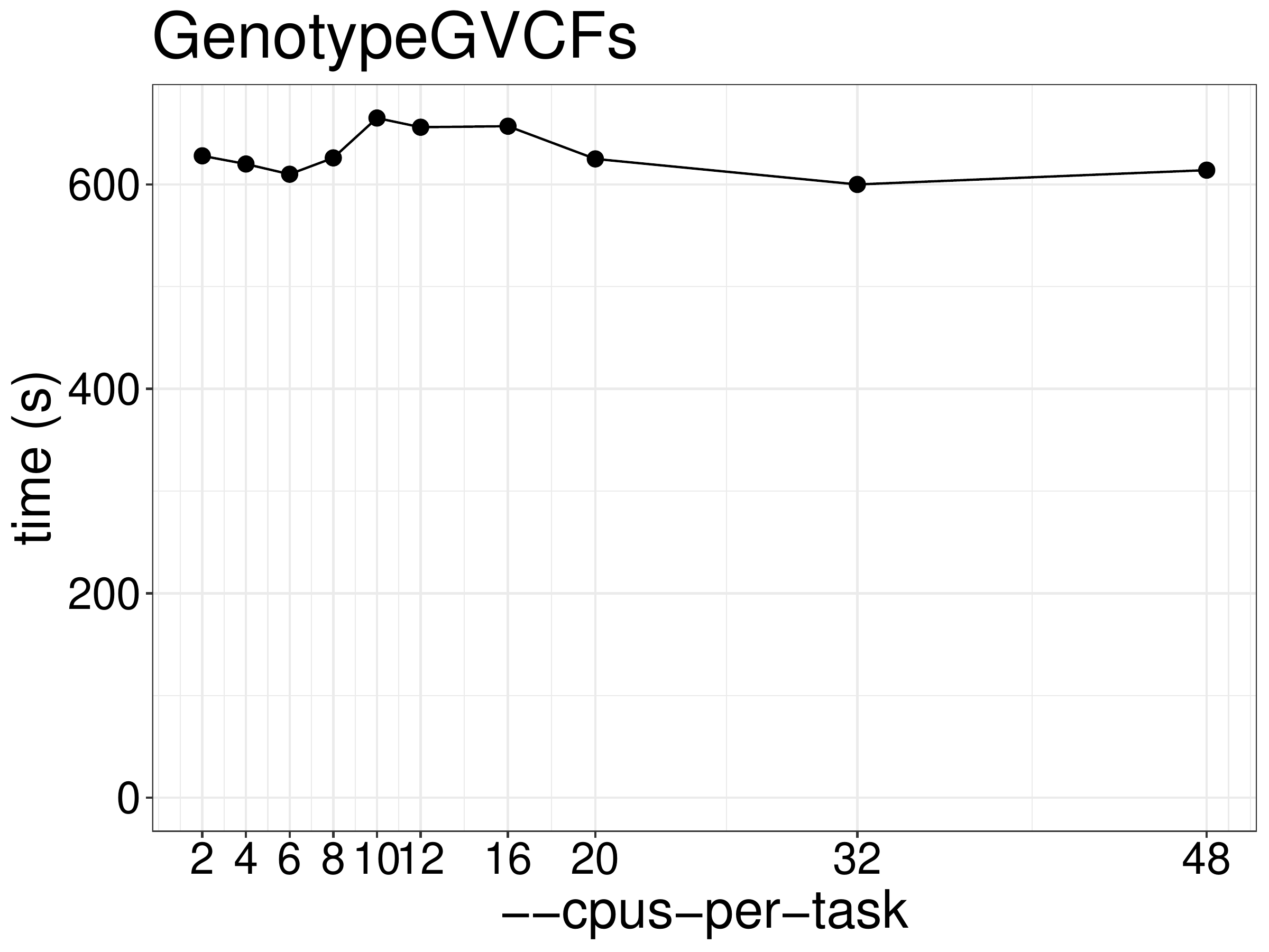
Figure 7.1: Runtime of GenotypeGVCFs as a function of the number of threads
We normally recommend that jobs be run with 70%-80% efficiency. Figure
7.2 shows efficiency for GenotypeGVCFs calculated from the
runtimes above. Based on this test GenotypeGVCFs jobs should be run
with 2 threads. Parallelism for this step is done by processing different
regions of the genome concurrently.
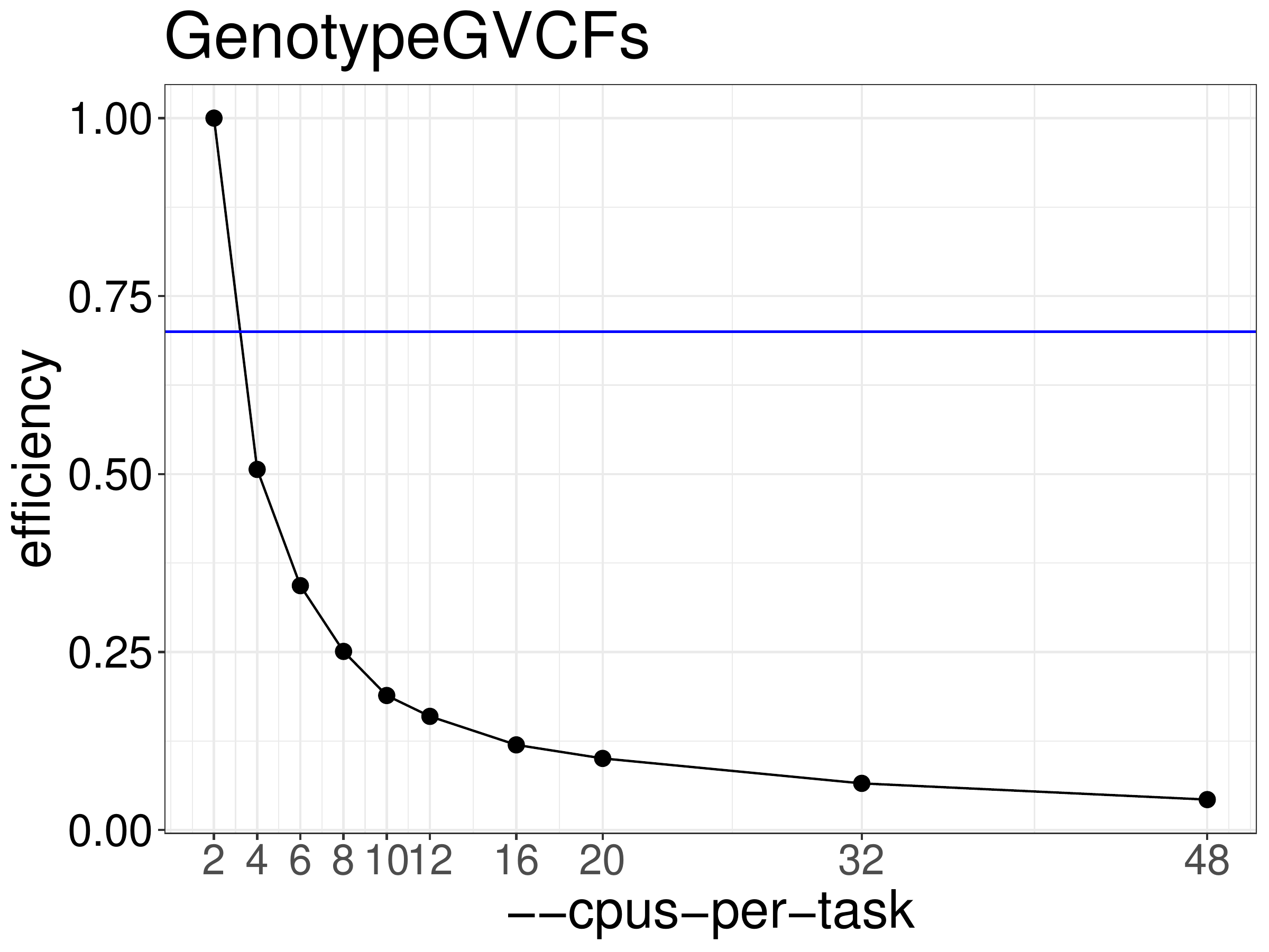
Figure 7.2: Efficiency of GenotypeGVCFs as a function of the number of threads
As for memory, increasing memory didn’t improve performance (figue 7.3).
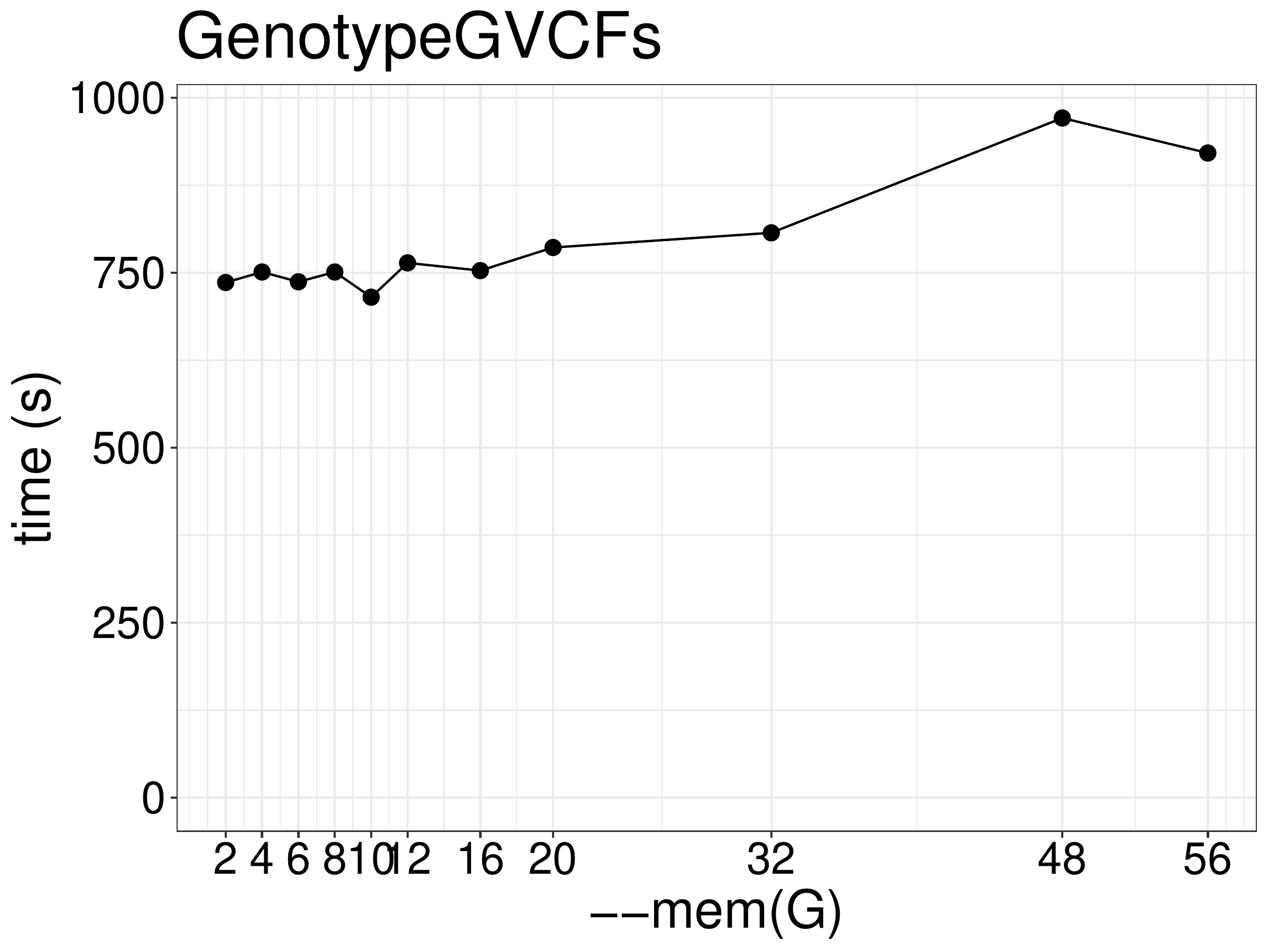
Figure 7.3: Runtime of GenotypeGVCFs as a function of memory
7.3 Optimized script
Example of running GenotypeGVCFs per chromosome:
cd data/; \
gatk --java-options "-Djava.io.tmpdir=/lscratch/$SLURM_JOBID -Xms2G -Xmx2G -XX:ParallelGCThreads=2" GenotypeGVCFs \
-R /fdb/igenomes/Homo_sapiens/UCSC/hg38/Sequence/WholeGenomeFasta/genome.fa \
-V gendb://chr1_gdb -O chr1.vcf.gz
cd data/; \
gatk --java-options "-Djava.io.tmpdir=/lscratch/$SLURM_JOBID -Xms2G -Xmx2G -XX:ParallelGCThreads=2" GenotypeGVCFs \
-R /fdb/igenomes/Homo_sapiens/UCSC/hg38/Sequence/WholeGenomeFasta/genome.fa \
-V gendb://chr2_gdb -O chr2.vcf.gzJob submission using swarm:
swarm -t 2 -g 2 -—gres=lscratch:100 --time=1:00:00 -m GATK/4.3.0.0 -f 07-GATK_GenotypeGVCFs.shNotes:
- The VCF files from each chromosome should be merged to one compressed file before next step, here we use
picardmodule to do the merge:
#!/bin/bash
module load picard
cd data/;
java -jar $PICARDJARPATH/picard.jar GatherVcfs I=chr1.vcf.gz I=chr2.vcf.gz I=chr3.vcf.gz I=chr4.vcf.gz I=chr5.vcf.gz I
=chr6.vcf.gz I=chr7.vcf.gz I=chr8.vcf.gz I=chr9.vcf.gz I=chr10.vcf.gz I=chr11.vcf.gz I=chr12.vcf.gz I=chr13.vcf.gz I=chr14.vcf.gz I
=chr15.vcf.gz I=chr16.vcf.gz I=chr17.vcf.gz I=chr18.vcf.gz I=chr19.vcf.gz I=chr20.vcf.gz I=chr21.vcf.gz I=chr22.vcf.gz I=chrX.vc
f I=chrY.vcf.gz I=chrM.vcf.gz O=merged.vcf.gzJob submission:
sbatch --cpus-per-task=2 --mem=2G --time=1:00:00 07-picard_GatherVcfs.sh