Chapter 5 HaplotypeCaller
5.1 Brief introduction
HaplotypeCaller is used to call potential variant sites per sample and save
results in GVCF format. With GVCF, it provides variant sites, and groups non-variant sites into blocks during the
calling process based on genotype quality. This is a way of compressing the
VCF file without losing any sites in order to do joint analysis in subsequent steps. The HaplotypeCaller
calculates the likelihoods for each record as homozygous-reference
or not based on the reference model.
5.2 Benchmarks
We did a benchmark on the performance of HaplotypeCaller with different
numbers of CPUs and memory. As show in figure 5.1, the runtime did not
reduce much given more threads for what is an essentially single-threaded
tool only using additional threads for garbage collection. The Spark based (parallel)
haplotype caller is still considered beta.
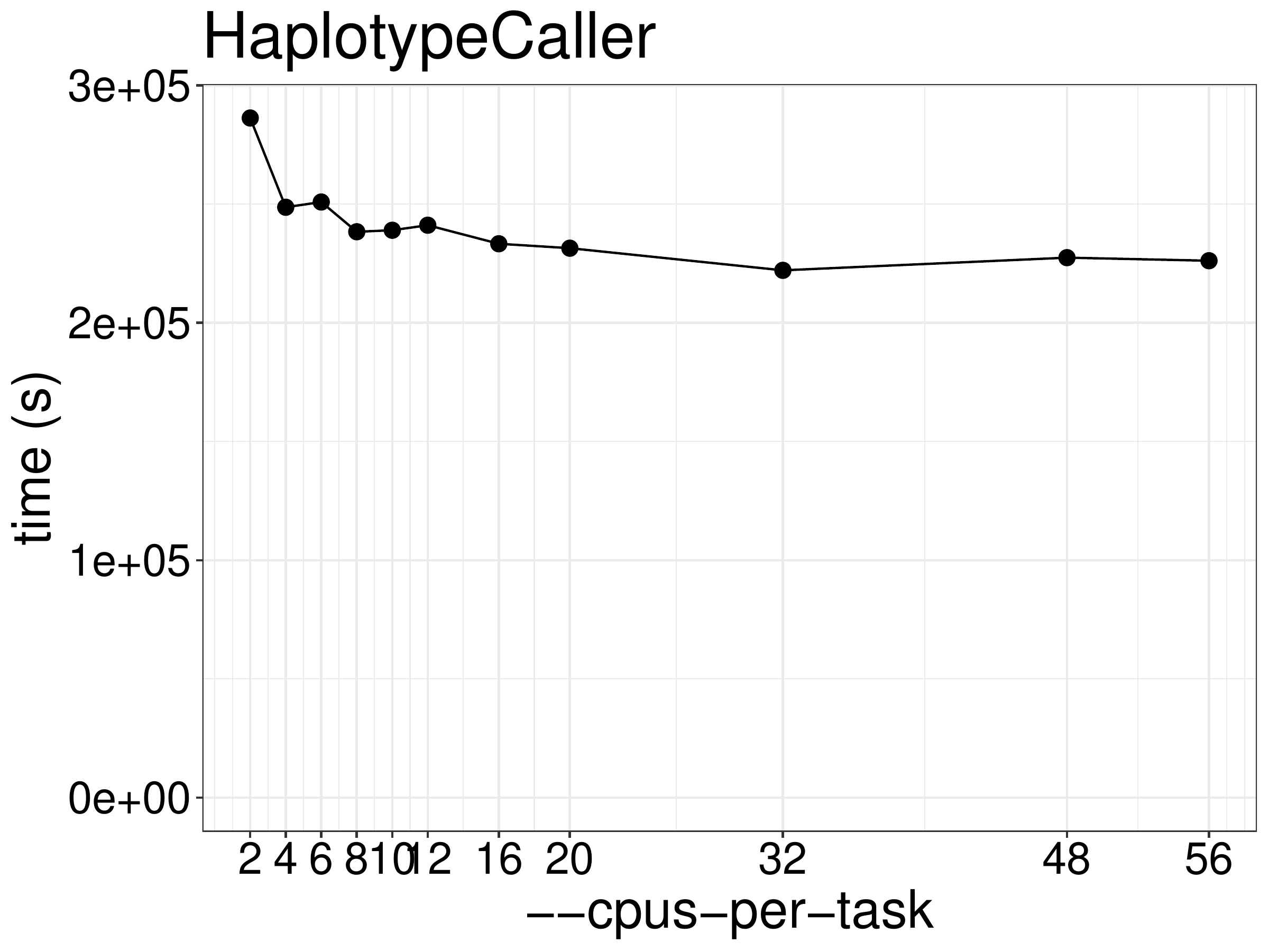
Figure 5.1: Runtime of HaplotypeCaller as a function of the number of threads
We normally recommend running jobs with 70%-80% efficiency. Based on figure
5.2), HaplotypeCaller should be run with no more than 2
threads.
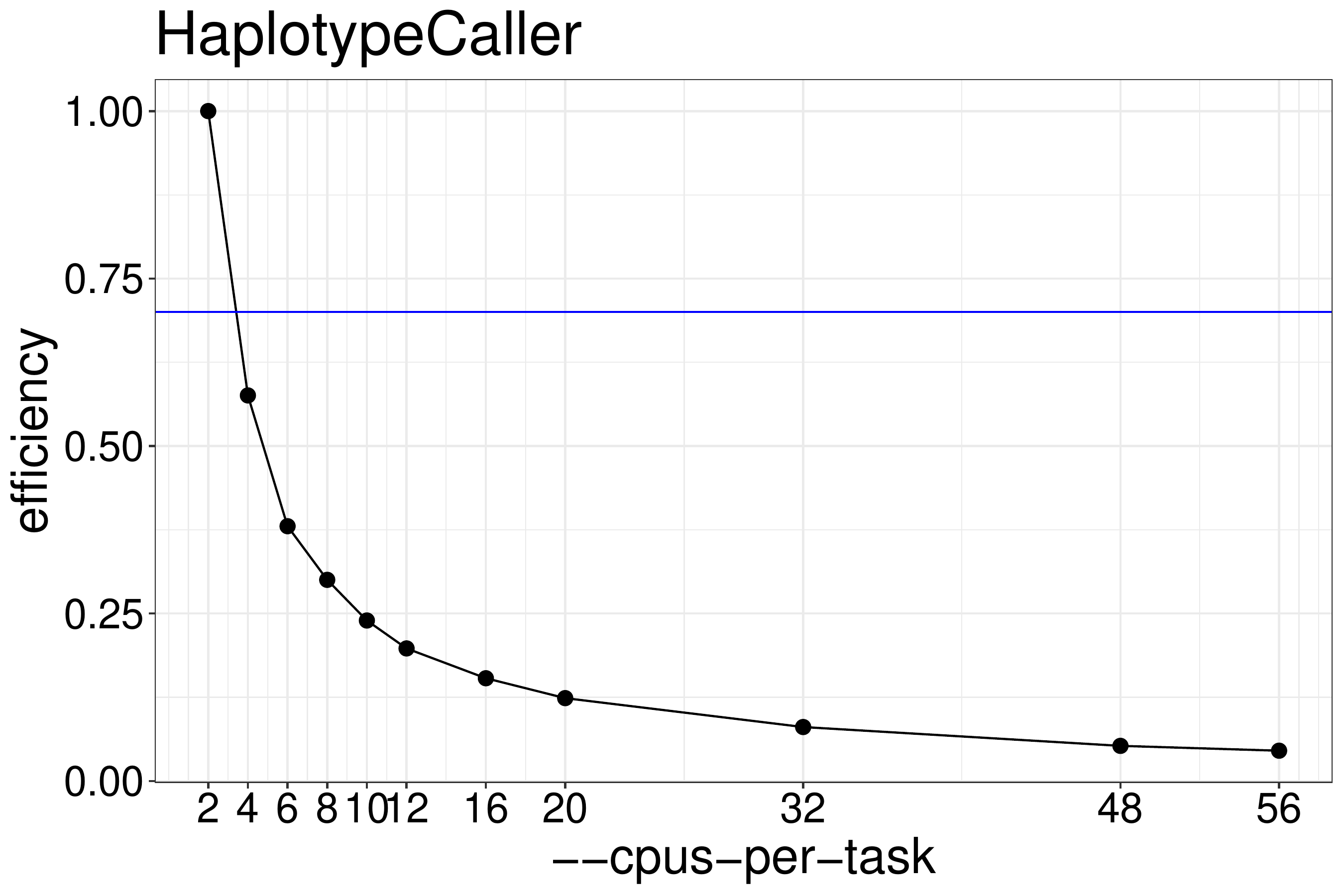
Figure 5.2: Efficiency of HaplotypeCaller as a function of the number of threads
As for memory, increasing memory didn’t improve performance (figue 5.3), although a minimum of 20GB memory is required for the job to finish with 2 threads.
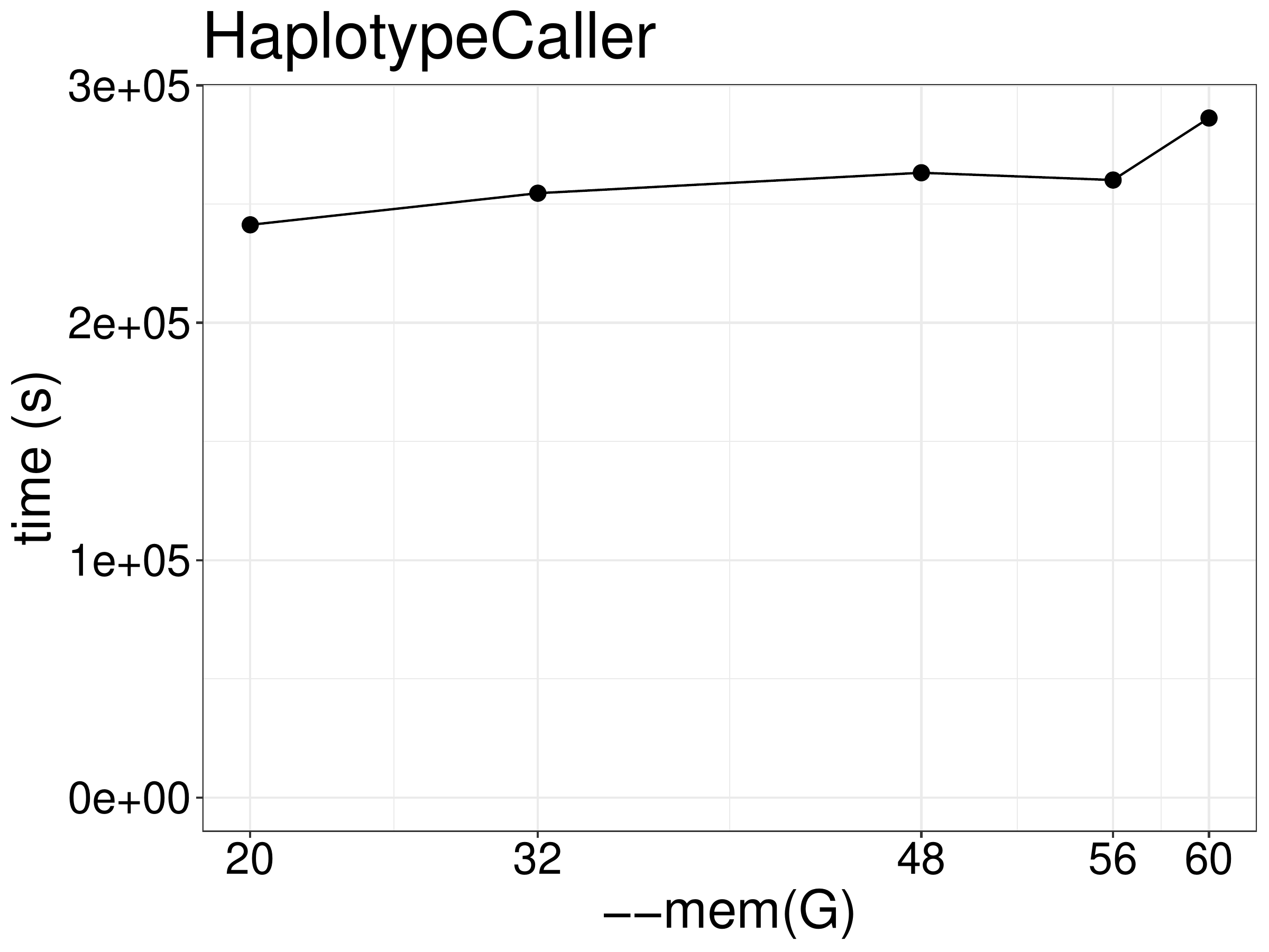
Figure 5.3: Runtime of HaplotypeCaller as a function of allocated memory
5.3 Optimized script
cd data/; \
gatk --java-options "-Djava.io.tmpdir=/lscratch/$SLURM_JOBID -Xms20G -Xmx20G -XX:ParallelGCThreads=2" HaplotypeCaller \
-R /fdb/igenomes/Homo_sapiens/UCSC/hg38/Sequence/WholeGenomeFasta/genome.fa \
-I NA12878_markdup_bqsr.bam \
-O NA12878.g.vcf.gz \
-ERC GVCF
cd data/; \
gatk --java-options "-Djava.io.tmpdir=/lscratch/$SLURM_JOBID -Xms20G -Xmx20G -XX:ParallelGCThreads=2" HaplotypeCaller \
-R /fdb/igenomes/Homo_sapiens/UCSC/hg38/Sequence/WholeGenomeFasta/genome.fa \
-I NA12891_markdup_bqsr.bam \
-O NA12891.g.vcf.gz \
-ERC GVCF
cd data/; \
gatk --java-options "-Djava.io.tmpdir=/lscratch/$SLURM_JOBID -Xms20G -Xmx20G -XX:ParallelGCThreads=2" HaplotypeCaller \
-R /fdb/igenomes/Homo_sapiens/UCSC/hg38/Sequence/WholeGenomeFasta/genome.fa \
-I NA12892_markdup_bqsr.bam \
-O NA12892.g.vcf.gz \
-ERC GVCFCommands can be submitted as separate jobs with swarm:
swarm -t 2 -g 20 —gres=lscratch:400 —time=80:00:00 -m GATK/4.3.0.0 -f 05-GATK_HaplotypeCaller.shNotes:
HaplotypeCalleris the most time consuming step.- Different from VCF, GVCF contains blocks with no evidence of variation.
- There are two types of GVCFs,
GVCFis recommended overBP_RESOLUTION(every site is kept). - We didn’t test using
HaplotypeCallerSparkbecause the Spark version did not match the output ofHaplotypeCaller. It is still in development and not recommended for production work.