Chapter 4 BQSR
4.1 Brief introduction
Systematic bias can originate from library preparation, sequencing,
manufacturing defects in the flowcell chips, sequencer variation, and sequencing
chemistry. It results in over- or underestimation of quality scores. GATK
recalibrates base quality scores by building an error model using known
covariates from all base calls (BaseRecalibrator), then applying adjustment to the
dataset based on the model (ApplyBQSR).
4.2 Benchmarks of BaseRecalibrator
We did a benchmark on the performance of BaseRecalibrator with different CPUs
and memory allocation. As shown in figure 4.1, the running time is not
reduced much when using more than 2 threads. This tool is not based on Spark so
any additional threads are only used for garbage collection.
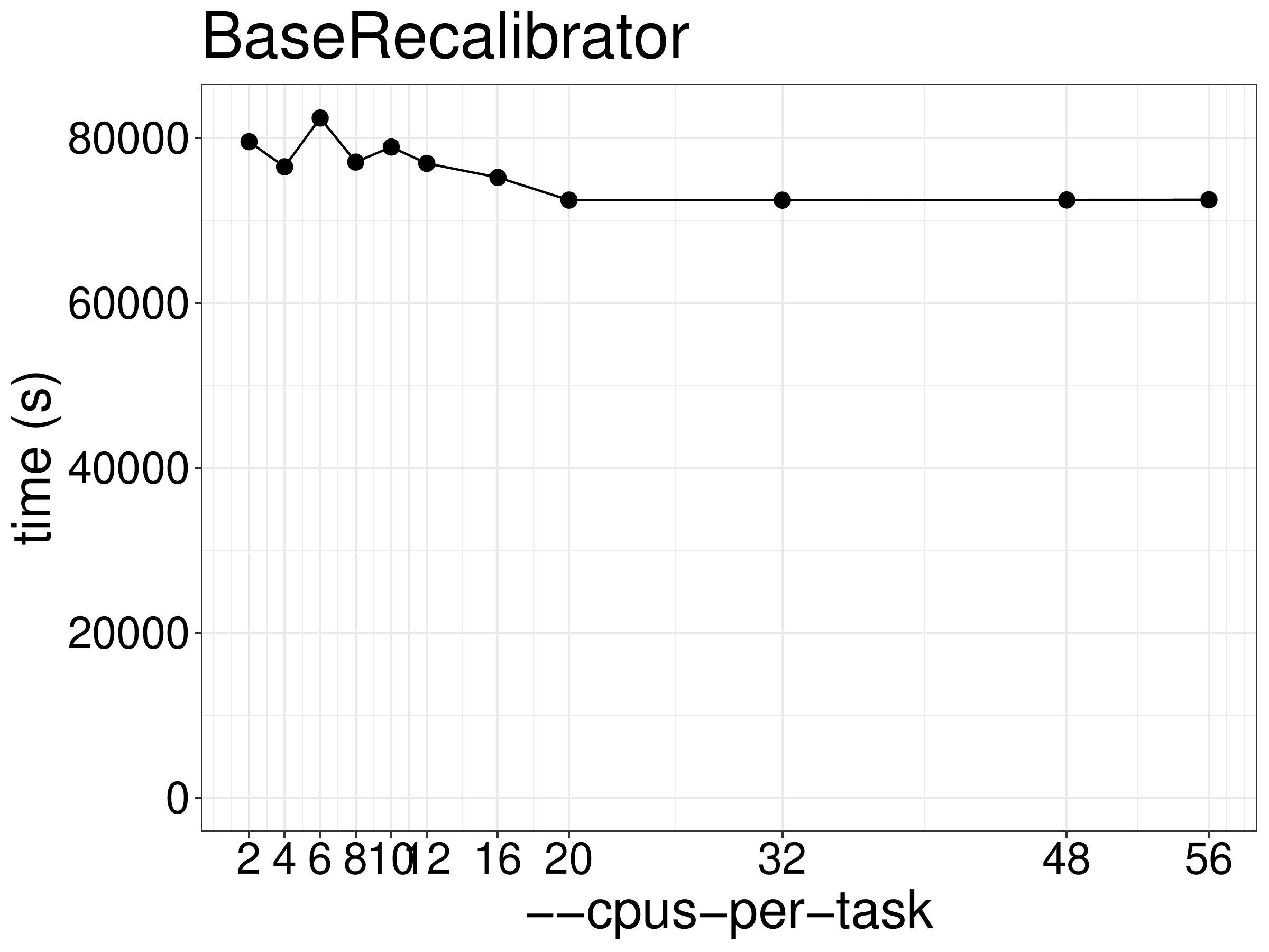
Figure 4.1: BaseRecalibrator runtime as a function of the number of threads.
We normally recommend to run scripts with 70%-80% efficiency. Based on the
results shown in figure 4.2, BaseRecalibrator should be run
with no more than 2 threads.
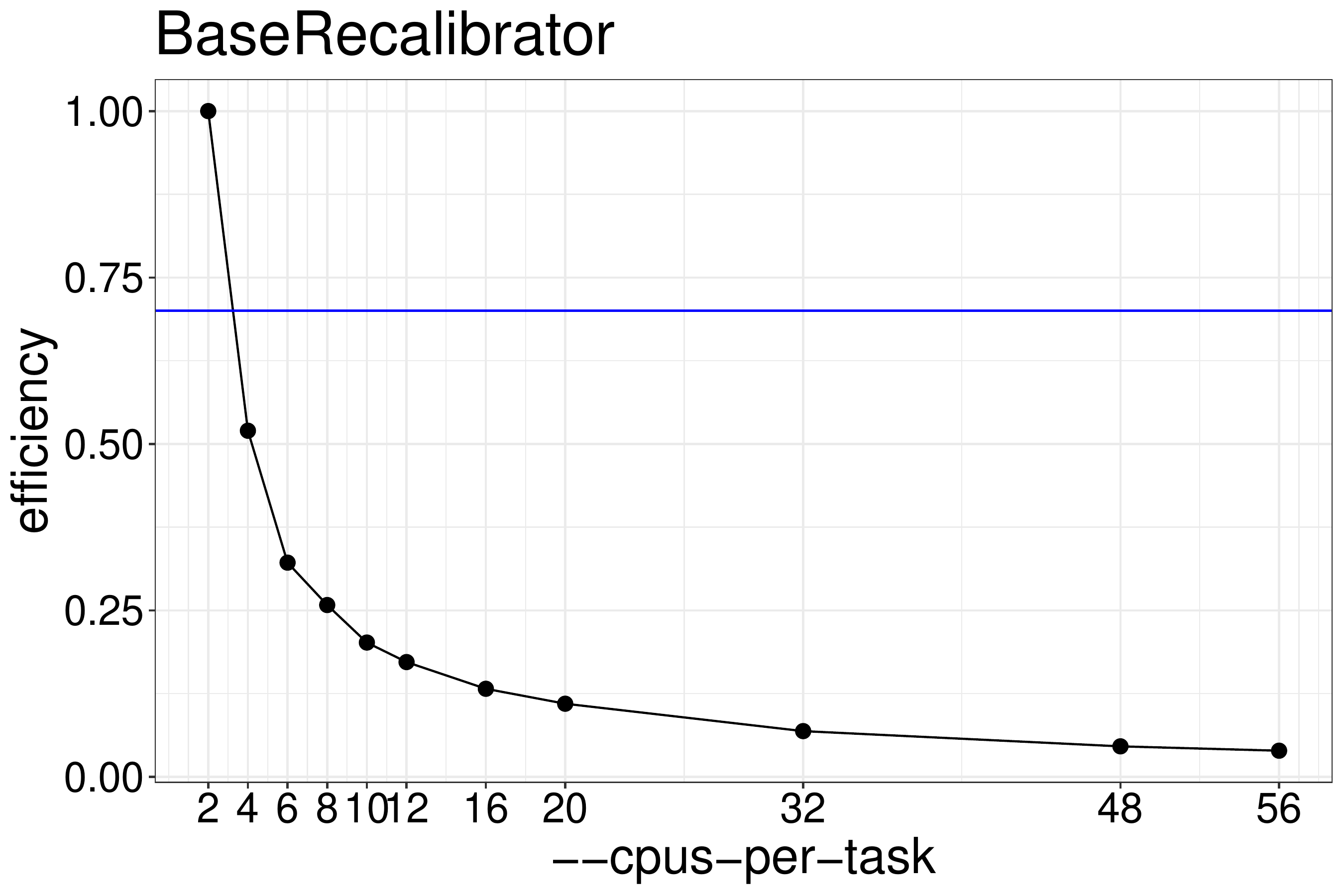
Figure 4.2: BaseRecalibrator efficiency calculated from runtimes
Increasing memory didn’t improve the performance beyond the minimal amount of memory (4GB at 2 threads) required to finish (figue 4.3).
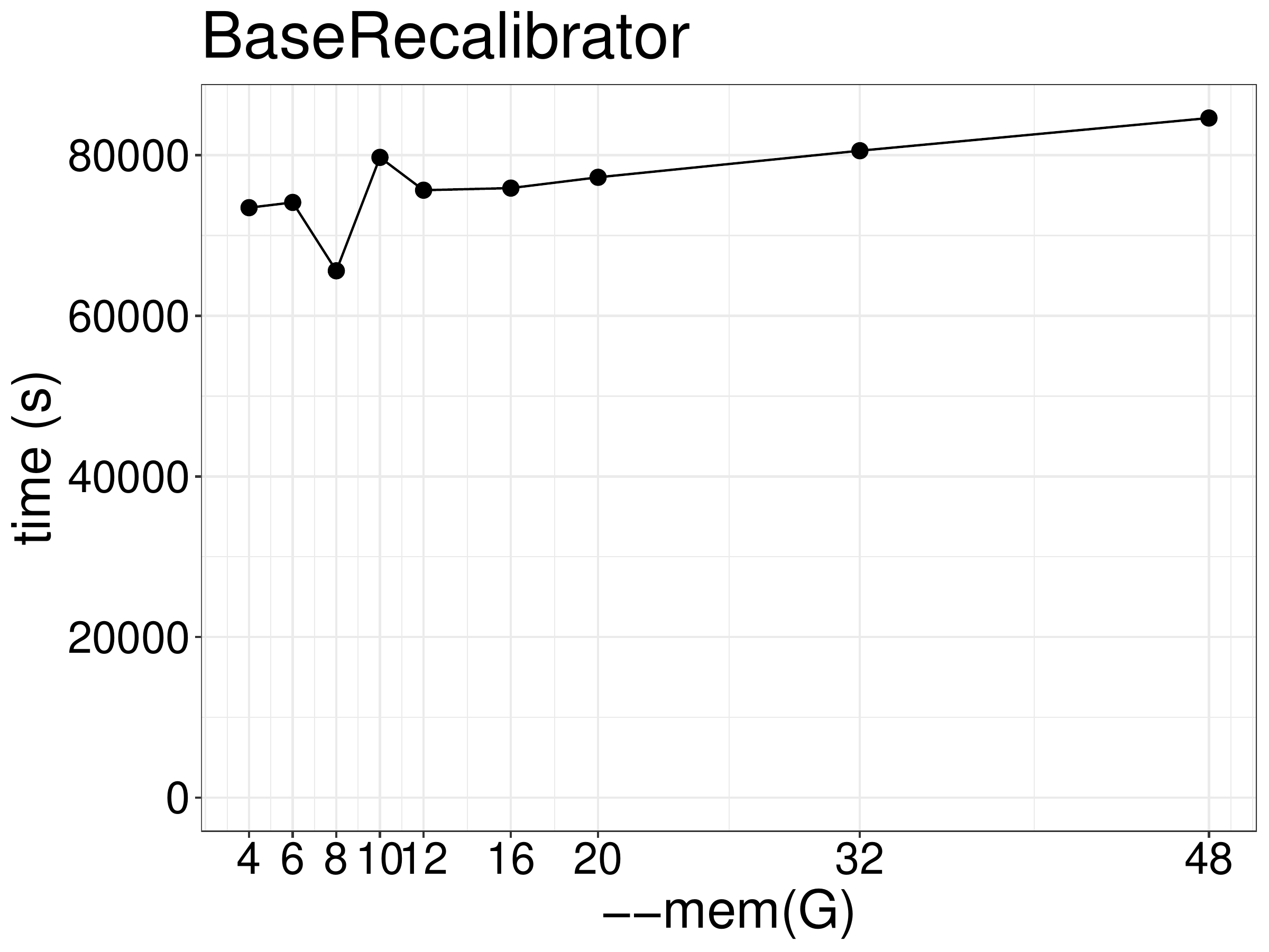
Figure 4.3: BaseRecalibrator runtime as a function of memory
4.3 Optimized script for BaseRecalibrator
Here is an example for building the model on each library:
cd data/; \
gatk --java-options "-Djava.io.tmpdir=/lscratch/$SLURM_JOBID -Xms4G -Xmx4G -XX:ParallelGCThreads=2" BaseRecalibrator \
-I NA12878_markdup.bam \
-R /fdb/igenomes/Homo_sapiens/UCSC/hg38/Sequence/WholeGenomeFasta/genome.fa \
-O NA12878_markdup_bqsr.report \
--known-sites /fdb/GATK_resource_bundle/hg38/dbsnp_146.hg38.vcf.gz \
--known-sites /fdb/GATK_resource_bundle/hg38/Homo_sapiens_assembly38.known_indels.vcf.gz \
--known-sites /fdb/GATK_resource_bundle/hg38/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz
cd data/; \
gatk --java-options "-Djava.io.tmpdir=/lscratch/$SLURM_JOBID -Xms4G -Xmx4G -XX:ParallelGCThreads=2" BaseRecalibrator \
-I NA12891_markdup.bam \
-R /fdb/igenomes/Homo_sapiens/UCSC/hg38/Sequence/WholeGenomeFasta/genome.fa \
-O NA12891_markdup_bqsr.report \
--known-sites /fdb/GATK_resource_bundle/hg38/dbsnp_146.hg38.vcf.gz \
--known-sites /fdb/GATK_resource_bundle/hg38/Homo_sapiens_assembly38.known_indels.vcf.gz \
--known-sites /fdb/GATK_resource_bundle/hg38/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz
cd data/; \
gatk --java-options "-Djava.io.tmpdir=/lscratch/$SLURM_JOBID -Xms4G -Xmx4G -XX:ParallelGCThreads=2" BaseRecalibrator \
-I NA12892_markdup.bam \
-R /fdb/igenomes/Homo_sapiens/UCSC/hg38/Sequence/WholeGenomeFasta/genome.fa \
-O NA12892_markdup_bqsr.report \
--known-sites /fdb/GATK_resource_bundle/hg38/dbsnp_146.hg38.vcf.gz \
--known-sites /fdb/GATK_resource_bundle/hg38/Homo_sapiens_assembly38.known_indels.vcf.gz \
--known-sites /fdb/GATK_resource_bundle/hg38/Mills_and_1000G_gold_standard.indels.hg38.vcf.gzThe known sites are used to mask out positions with known variation to avoid confusing real variation with errors.
Commands can be submitted as separate jobs using swarm:
swarm -t 2 -g 4 --gres=lscratch:400 --time=24:00:00 -m GATK/4.3.0.0 -f 04-GATK_BaseRecalibrator.sh4.4 Benchmarks of ApplyBQSR
We did a benchmark on the performance of ApplyBQSR with different CPUs and
memory allocation. As show in figure 4.4, the running time is not reduce much when using more than 2 threads. This tool is not based on Spark so any additional threads are only used for garbage collection.
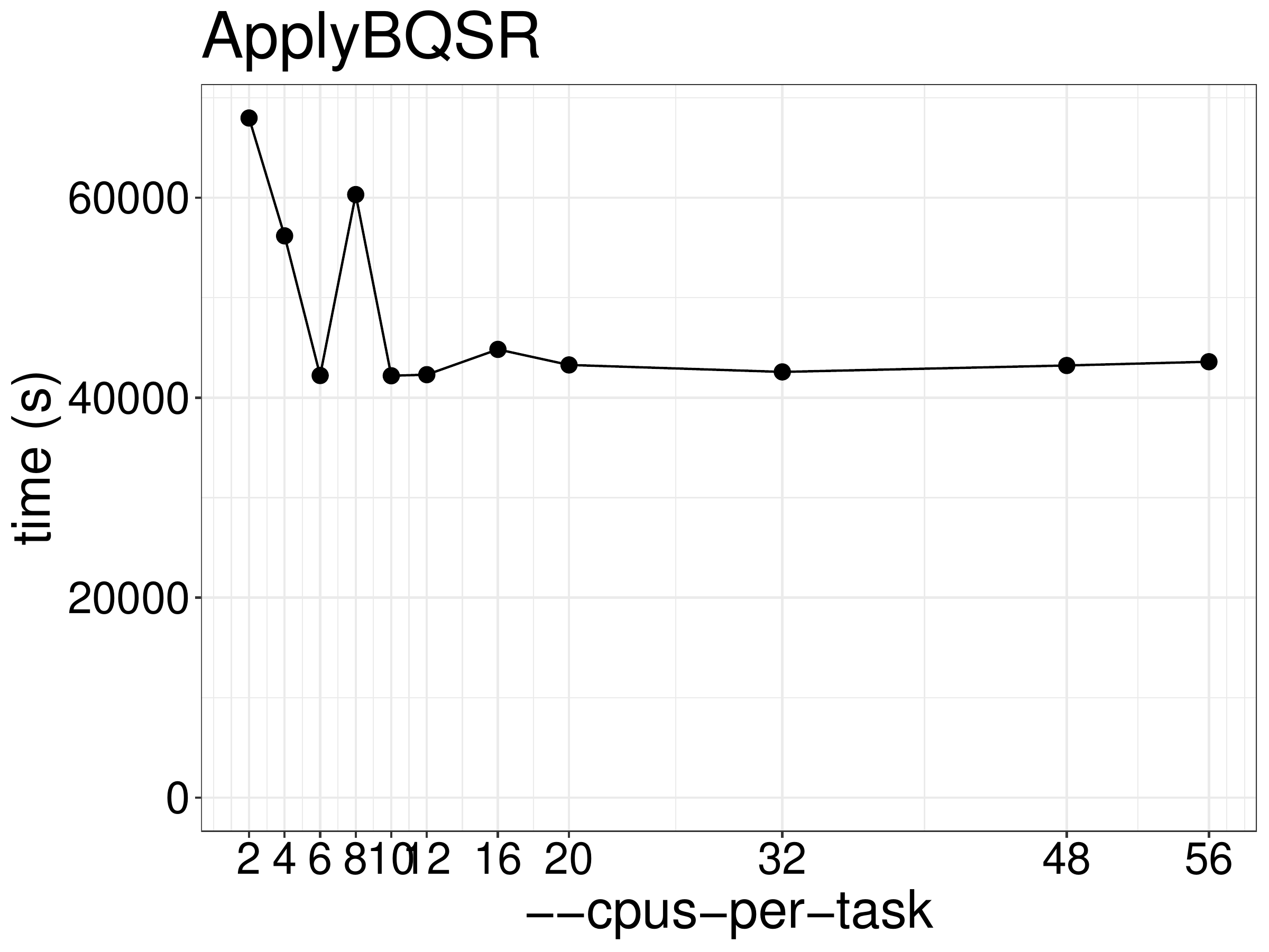
Figure 4.4: ApplyBQSR runtime as a function of the number of threads.
We normally recommend to run jobs with 70%-80% efficiency. Based on the
results shown in figure 4.5 ApplyBQSR should be run with no
more than 2 threads.
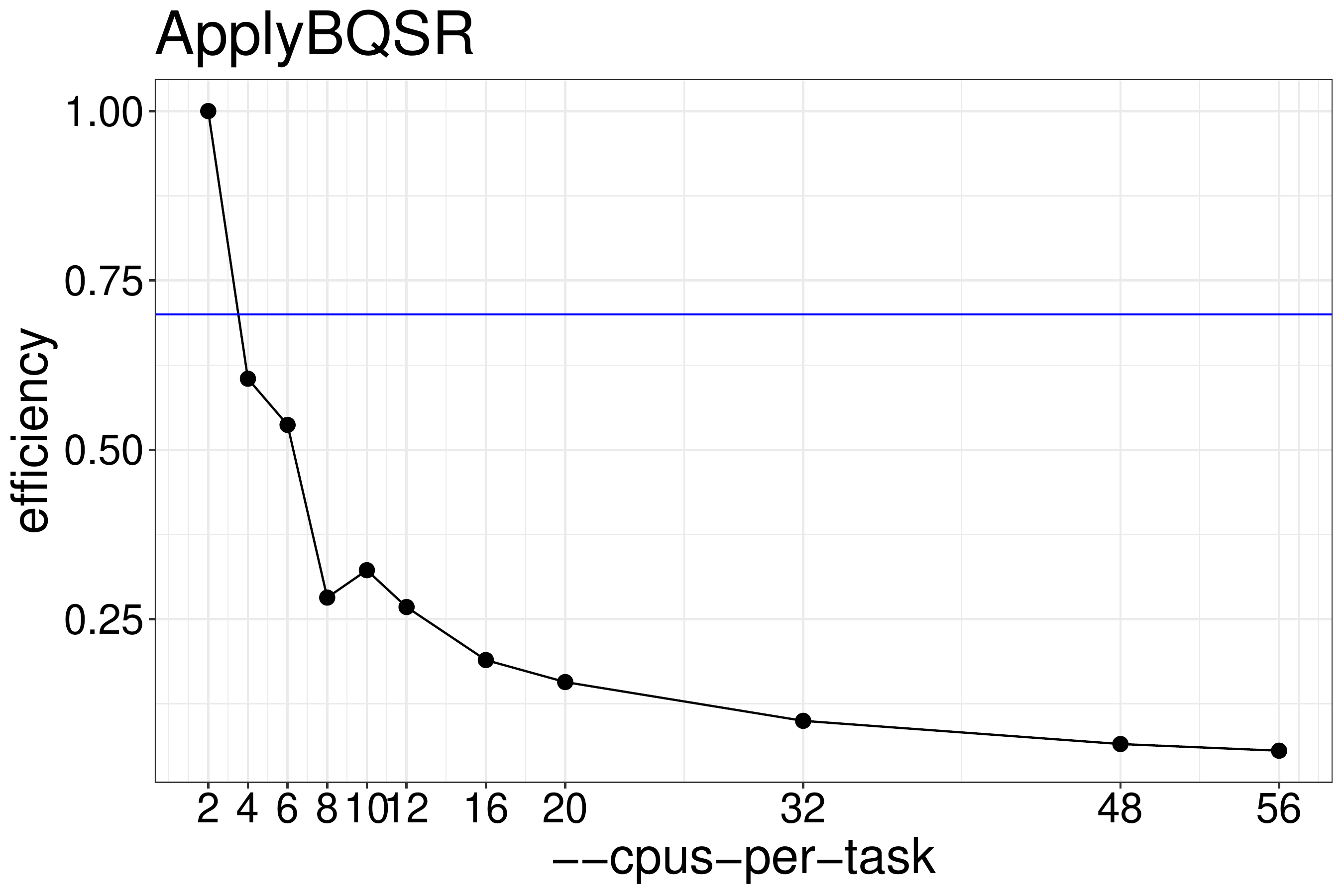
Figure 4.5: ApplyBQSR efficiency calculated from runtimes
Increasing memory didn’t improve the performance.(figure 4.6).
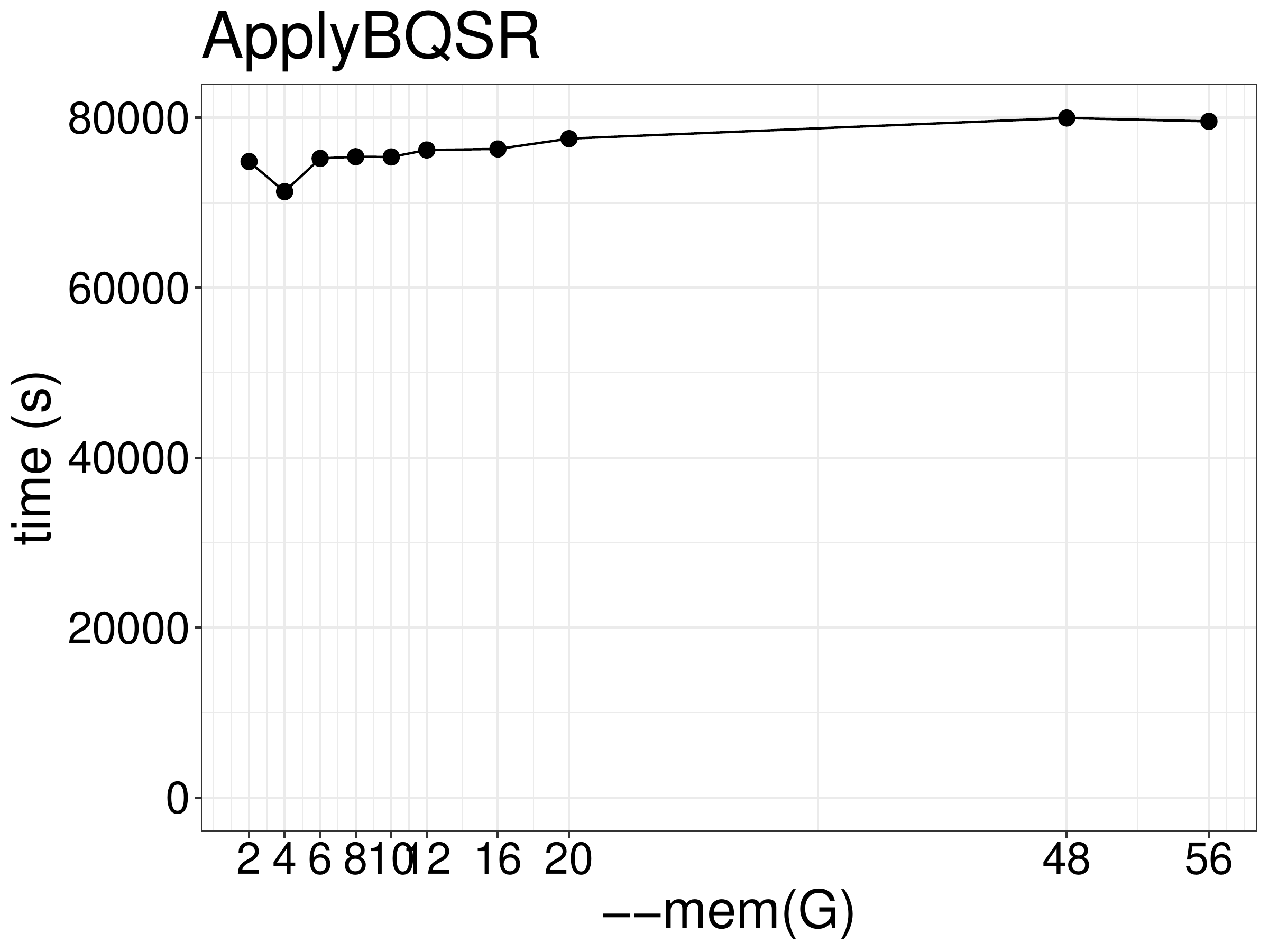
Figure 4.6: ApplyBQSR runtime as a function of memory
4.5 Optimized script for ApplyBQSR
We apply the recalibration model to each sample:
cd data/; \
gatk --java-options "-Djava.io.tmpdir=/lscratch/$SLURM_JOBID -Xms2G -Xmx2G -XX:ParallelGCThreads=2" ApplyBQSR \
-I NA12878_markdup.bam \
-R /fdb/igenomes/Homo_sapiens/UCSC/hg38/Sequence/WholeGenomeFasta/genome.fa \
--bqsr-recal-file NA12878_markdup_bqsr.report \
-O NA12878_markdup_bqsr.bam
cd data/; \
gatk --java-options "-Djava.io.tmpdir=/lscratch/$SLURM_JOBID -Xms2G -Xmx2G -XX:ParallelGCThreads=2" ApplyBQSR \
-I NA12891_markdup.bam \
-R /fdb/igenomes/Homo_sapiens/UCSC/hg38/Sequence/WholeGenomeFasta/genome.fa \
--bqsr-recal-file NA12891_markdup_bqsr.report \
-O NA12891_markdup_bqsr.bam
cd data/; \
gatk --java-options "-Djava.io.tmpdir=/lscratch/$SLURM_JOBID -Xms2G -Xmx2G -XX:ParallelGCThreads=2" ApplyBQSR \
-I NA12892_markdup.bam \
-R /fdb/igenomes/Homo_sapiens/UCSC/hg38/Sequence/WholeGenomeFasta/genome.fa \
--bqsr-recal-file NA12892_markdup_bqsr.report \
-O NA12892_markdup_bqsr.bamJob submission on Biowulf using swarm:
swarm -t 2 -g 2 --gres=lscratch:400 --time=24:00:00 -m GATK/4.3.0.0 -f 04-GATK_ApplyBQSR.shNote:
- BQSR is read group specific.
BQSRPipelineSparkis not discussed here since it is a BETA tool and is not yet ready for use in production.