
The PHENIX software suite is a highly automated system for macromolecular structure determination that can rapidly arrive at an initial partial model of a structure without significant human intervention, given moderate resolution and good quality data.
This application requires a graphical connection.
Allocate an HPC OnDemand graphical session, create a terminal window, and run the program.
Sample session (user input in bold):
[user@cn0000 ~]$ module load Phenix [user@cn0000 ~]$ phenix & ... [user@cn0000 ~]$ exit
Create a batch input file (e.g. Phenix.sh). For example:
#!/bin/bash module load Phenix phenix.autobuild seq_file=p9.seq data=p9-solve.mtz \ input_map_file=p9-resolve.mtz resolution=2.4 \ ncs_copies=1 nproc=$SLURM_CPUS_ON_NODE \ temp_dir=/lscratch/$SLURM_JOB_ID
Submit this job using the Slurm sbatch command.
sbatch [--cpus-per-task=#] [--mem=#] --gres=lscratch:50 Phenix.sh
Create a swarmfile (e.g. Phenix.swarm). For example:
phenix.elbow input_1.pdb phenix.elbow input_2.pdb phenix.elbow input_3.pdb phenix.elbow input_4.pdb
Submit this job using the swarm command.
swarm -f Phenix.swarm [-g #] [-t #] --module Phenixwhere
| -g # | Number of Gigabytes of memory required for each process (1 line in the swarm command file) |
| -t # | Number of threads/CPUs required for each process (1 line in the swarm command file). |
| --module Phenix | Loads the Phenix module for each subjob in the swarm |
Rosetta has been compiled specifically for Phenix, and is available via two environment variables PHENIX_ROSETTA_PATH and ROSETTA3_DB. Do NOT load the Rosetta modules built for Biowulf, as these are NOT compiled specifically for Phenix. Also note that structural models are limited to standard amino acids and other limitations.
Here is an example of structural refinement using Rosetta to generate structural models (this file is named Rosetta_refine.sh):
#!/bin/bash #SBATCH --cpus-per-task=4 #SBATCH --time=72:00:00 #SBATCH --mem=50g #SBATCH --gres=lscratch:50 module load Phenix phenix.rosetta_refine input.pdb input.mtz \ nproc=$SLURM_CPUS_ON_NODE \ temp_dir=/lscratch/$SLURM_JOB_ID \ post_refine=True
Then submit to the cluster:
sbatch Rosetta_refine.sh
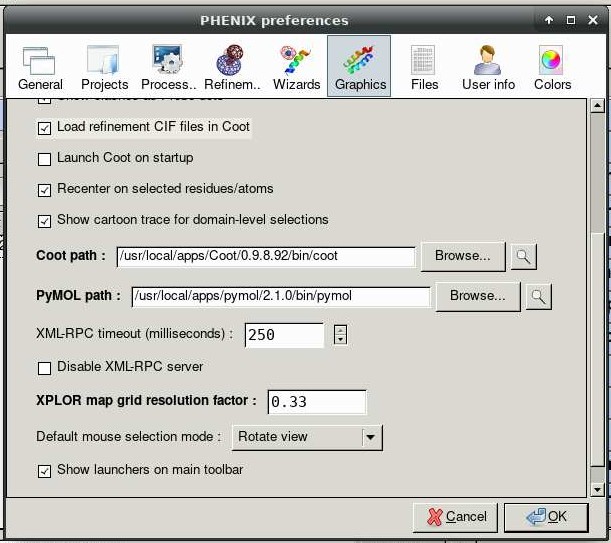
Phenix can launch graphical applications Coot and Pymol. Navigate to Preferences -> Graphics and insert this string into the text box for PyMOL path:
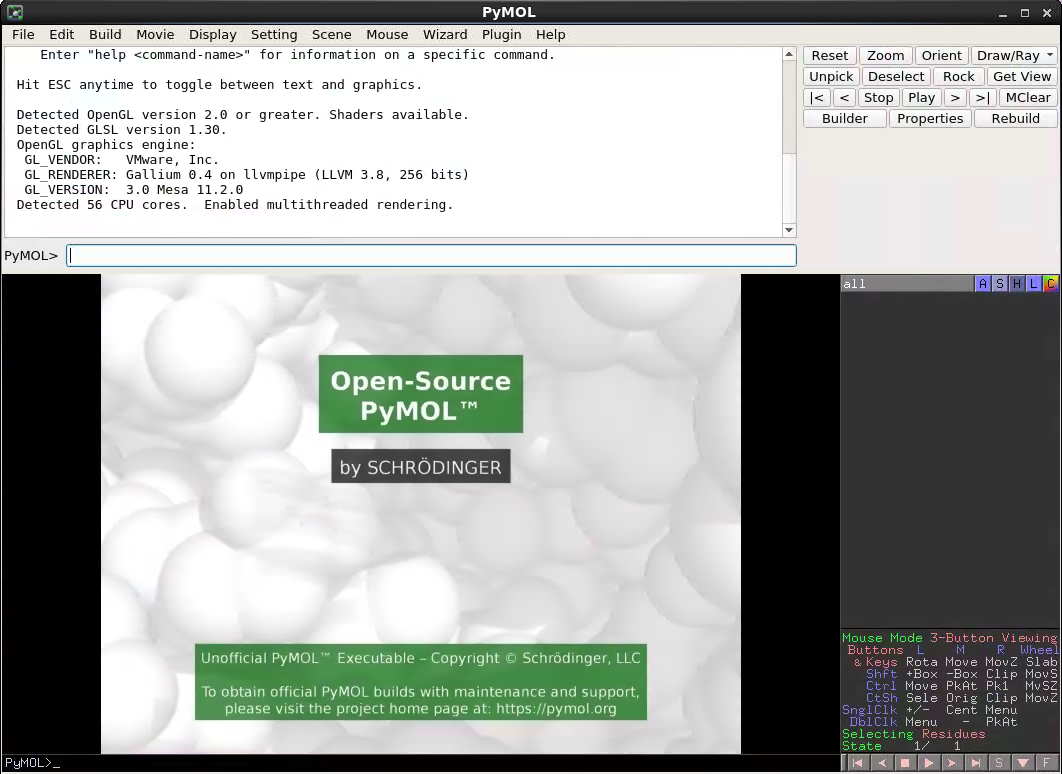
Now when the PyMOL button is clicked, you should see this:
The CryoFit protocol can be used to fit a fasta or pdb into a cryoEM map.
CryoFit can be both in the GUI and from the commandline. Here is how to run the tutorials from the command line:
cp $PHENIX_MODULES_DIR/cryo_fit/tutorial_input_files/*.{mrc,pdb} .
phenix.cryo_fit GTPase_activation_center_tutorial.pdb GTPase_activation_center_tutorial_gaussian_1p5.mrc