Stan is a state-of-the-art platform for statistical modeling and high-performance statistical computation. Thousands of users rely on Stan for statistical modeling, data analysis, and prediction in the social, biological, and physical sciences, engineering, and business.
-- from https://mc-stan.org/
cmdstan is the command line interface to stan. Because it is meant to be used from within its own source tree, Biowulf provides a helper script which assists users in compiling their own local version of cmdstan as well as an example model, data, and batch script for illustration.
Stan models have to be explicitly written to use within-chain parallelism. See the stan documentation.
R and python include stan interfaces as well.
cmdstan assumes that it is run from within the source tree it was buit in. To make it simple to use, we provide a wrapper script that builds a local copy of cmdstan with a given compiler/MPI combination. The script along with source tar balls are the only thing provided by the module.
As a first step, create a cmdstan build:
[user@biowulf]$ module load cmdstan [+] Loading gcc 11.3.0 [+] Loading cmdstan 2.30.1 [user@biowulf]$ cmdstan help NAME cmdstan -- cmdstan installation wrapper SYNOPSIS cmdstanCOMMANDS help show this help message compile [--mpi|--nompi] submit a compile job. Deaults to --mpi example [--nobuild] copy example to local stan directory and build it (unless the --nobuild is option is used) DESCRIPTION The `cmdstan` wrapper script will submit a batch job to build cmdstan in the current directory. You will be notified by mail when the build is finished. It alows building either the default build or an MPI-based build. VERSIONS cmdstan 2.30.1 gcc 11.3.0 mpi openmpi/4.1.3/gcc-11.3.0 arch x2680 [user@biowulf]$ cmdstan compile Submitted compile job 53209944 You will receive an email when compile is complete
This will submit a batch job compiling cmdstan in the current
working directory. It will use the processor architecture, compiler,
and mpi module indicated in the help message. Once the job is done,
a email notification will be sent. Use --nompi to build
stan to use TBB (thread) based parallelism for running within a node. In
either case models have to be written explicitly to enable within-chain
parallelism and between-chain parallelism may not function properly
for MPI. Once the build is done:
[user@biowulf]$ ls -lh drwxr-xr-x 10 user group 4096 Mar 24 09:00 cmdstan-2.21.0-mpi -rw-r--r-- 1 user group 126916 Mar 24 09:04 cmdstan-2.21.0-mpi.53209944.log [user@biowulf]$ cd cmdstan-2.30.1-mpi
Stan models have to be compiled into standalone executables from within this directory. The wrapper script can compile an example - again as a batch job because it requires the mpi module and needs to be compiled on an infiniband node
[user@biowulf]$ cmdstan example running from within compiled cmdstan home dir copying example one sec - compiling model as a batch job command run: module load openmpi/3.1.4/gcc-9.2.0; make linreg/linreg_par This command can be run from a batch job or sinteractive session where the openmpi library can be loaded. Submitted compile job 53210535 You will receive an email when compile is complete
This will create a linreg directory with a model,
the compiled model, as well as a batch script for submission. Let's run
the example model on a single node with 28 tasks:
[user@biowulf]$ cd linreg
[user@biowulf]$ cat batch.sh
#! /bin/bash
#SBATCH --ntasks=28
#SBATCH --ntasks-per-core=1
#SBATCH --constraint=x2680
#SBATCH --exclusive
#SBATCH --partition=multinode
#SBATCH --gres=lscratch:10
#SBATCH --mem-per-cpu=2g
module load openmpi/3.1.4/gcc-9.2.0
N=1000
n=500
S=$SLURM_NTASKS
O=4
fn=/lscratch/$SLURM_JOB_ID/dat_order_${O}_shards_${S}.Rdump
# copy the input file and set the number of shards
sed "s/shards <- S/shards <- $S/" \
dat_order_${O}_shards_S.Rdump > $fn
srun --mpi=pmix_v3 ./linreg_par \
sample num_samples=$N num_warmup=$n \
data file=$fn \
output file=output_order_${O}_shards_${S}.csv
[user@biowulf]$ sbatch batch.sh
53161946
[user@biowulf]$ sleep 15m
[user@biowulf]$ cat slurm-53161946.out
...snip...
Gradient evaluation took 0.07 seconds
1000 transitions using 10 leapfrog steps per transition would take 700 seconds.
Adjust your expectations accordingly!
Iteration: 1 / 1500 [ 0%] (Warmup)
Iteration: 100 / 1500 [ 6%] (Warmup)
Iteration: 200 / 1500 [ 13%] (Warmup)
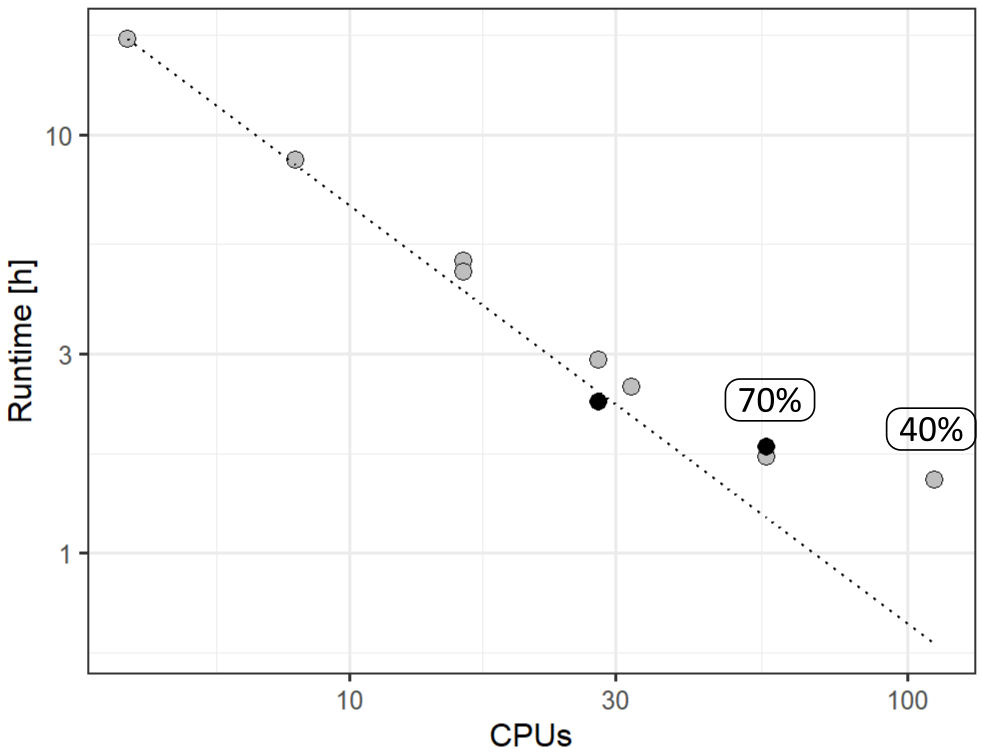
This model scales well up to 16 tasks (32 CPUs since we set
--ntasks-per-core=1 in this example) and still runs at
about 70% efficiency with 28 tasks (56 CPUs). In addition, it appears
to do fairly well with 2 tasks per core. In the following graph,
runtime is shown as a function of the number of CPUs. Light grey
circles used 1 task per core. Black used 2 tasks per core. The line
shows ideal scaling.
Here is how you would compile your own model. We'll use the same example as above.
[user@biowulf]$ mkdir linreg2 [user@biowulf]$ cp linreg/linreg_par.stan linreg2 [user@biowulf]$ sinteractive --constraint=x2680 --mem=4g salloc.exe: Pending job allocation 46116226 salloc.exe: job 46116226 queued and waiting for resources salloc.exe: job 46116226 has been allocated resources salloc.exe: Granted job allocation 46116226 salloc.exe: Waiting for resource configuration salloc.exe: Nodes cn3144 are ready for job [user@cn3144]$ module load openmpi/3.1.4/gcc-9.2.0 [user@cn3144]$ make linreg2/linreg_par ... [user@cn3144]$ exit [user@biowulf]$