Flye is a de novo assembler for long and noisy reads, such as those produced by PacBio and Oxford Nanopore Technologies. The algorithm uses an A-Bruijn graph to find the overlaps between reads and does not require them to be error-corrected. After the initial assembly, Flye performs an extra repeat classification and analysis step to improve the structural accuracy of the resulting sequence. The package also includes a polisher module, which produces the final assembly of high nucleotide-level quality.
Flye replaces abruijn and does provide a abruijn script for
backwards compatibility.
A 5Mb bacterial genome with ~80x coverage was assembled on one of our compute nodes (6GB memory; 16 CPUs) in about 30min. A ~150 Mb D. melanogaster genome was assembled in 13h (100GB memory; 32 CPUs).
$FLYE_TEST_DATAAllocate an interactive session and run the program. Sample session:
Note that --genome-size is optional since version 2.8
[user@biowulf]$ sinteractive --mem=14g --cpus-per-task=16 --gres=lscratch:10
salloc.exe: Pending job allocation 46116226
salloc.exe: job 46116226 queued and waiting for resources
salloc.exe: job 46116226 has been allocated resources
salloc.exe: Granted job allocation 46116226
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn3144 are ready for job
[user@cn3144 ~]$ module load flye/2.9.5
[user@cn3144 ~]$ cd /lscratch/$SLURM_JOB_ID
[user@cn3144 ~]$ mkdir assembly_ecoli
[user@cn3144 ~]$ cp $FLYE_TEST_DATA/SRR1284073_gt10k.fasta.gz .
[user@cn3144 ~]$ flye -t $SLURM_CPUS_PER_TASK --pacbio-raw SRR1284073_gt10k.fasta.gz \
-o assembly_ecoli --genome-size 5m
[2024-09-25 20:17:21] INFO: Starting Flye 2.9.5-b1801
[2024-09-25 20:17:21] INFO: >>>STAGE: configure
[2024-09-25 20:17:21] INFO: Configuring run
[2024-09-25 20:17:23] INFO: Total read length: 424263329
[2024-09-25 20:17:23] INFO: Input genome size: 5000000
[2024-09-25 20:17:23] INFO: Estimated coverage: 84
[2024-09-25 20:17:23] INFO: Reads N50/N90: 17480 / 11579
[...snip...]
[2024-09-25 20:27:41] INFO: >>>STAGE: finalize
[2024-09-25 20:27:41] INFO: Assembly statistics:
Total length: 4775724
Fragments: 5
Fragments N50: 2914006
Largest frg: 2914006
Scaffolds: 0
Mean coverage: 65
[user@cn3144 ~]$ ll assembly_ecoli
total 9.4M
drwxr-xr-x 2 user group 4.0K Sep 25 20:20 00-assembly
drwxr-xr-x 2 user group 4.0K Sep 25 20:21 10-consensus
drwxr-xr-x 2 user group 4.0K Sep 25 20:23 20-repeat
drwxr-xr-x 2 user group 4.0K Sep 25 20:23 30-contigger
drwxr-xr-x 2 user group 4.0K Sep 25 20:27 40-polishing
-rw-r--r-- 1 user group 4.7M Sep 25 20:27 assembly.fasta
-rw-r--r-- 1 user group 4.6M Sep 25 20:27 assembly_graph.gfa
-rw-r--r-- 1 user group 2.6K Sep 25 20:27 assembly_graph.gv
-rw-r--r-- 1 user group 229 Sep 25 20:27 assembly_info.txt
-rw-r--r-- 1 user group 69K Sep 25 20:27 flye.log
-rw-r--r-- 1 user group 93 Sep 25 20:27 params.json
[user@cn3144 ~]$ # copy back to data
[user@cn3144 ~]$ cp -r assembly_ecoli /data/$USER/badbadproject
[user@cn3144 ~]$ exit
salloc.exe: Relinquishing job allocation 46116226
[user@biowulf ~]$
Create a batch input file (e.g. flye.sh) similar to the following:
#! /bin/bash ml flye/2.9.5 || exit 1 cd /lscratch/$SLURM_JOB_ID cp /data/users/some/where/reads.fa.gz . flye -t $SLURM_CPUS_PER_TASK --pacbio-raw reads.fasta.gz -o assembly_dmelanogaster --genome-size 150m mv assembly_dmelanogaster /data/$USER/badbadproject
This particular example made use of data set SRX499318 filtered to reads >14k length resulting in a 90x coverage of the ~150Mb D. melanogaster genome.
Submit this job using the Slurm sbatch command.
sbatch --mem=120g --cpus-per-task=32 --gres=lscratch:300 flye.batch --time=1-00:00:00
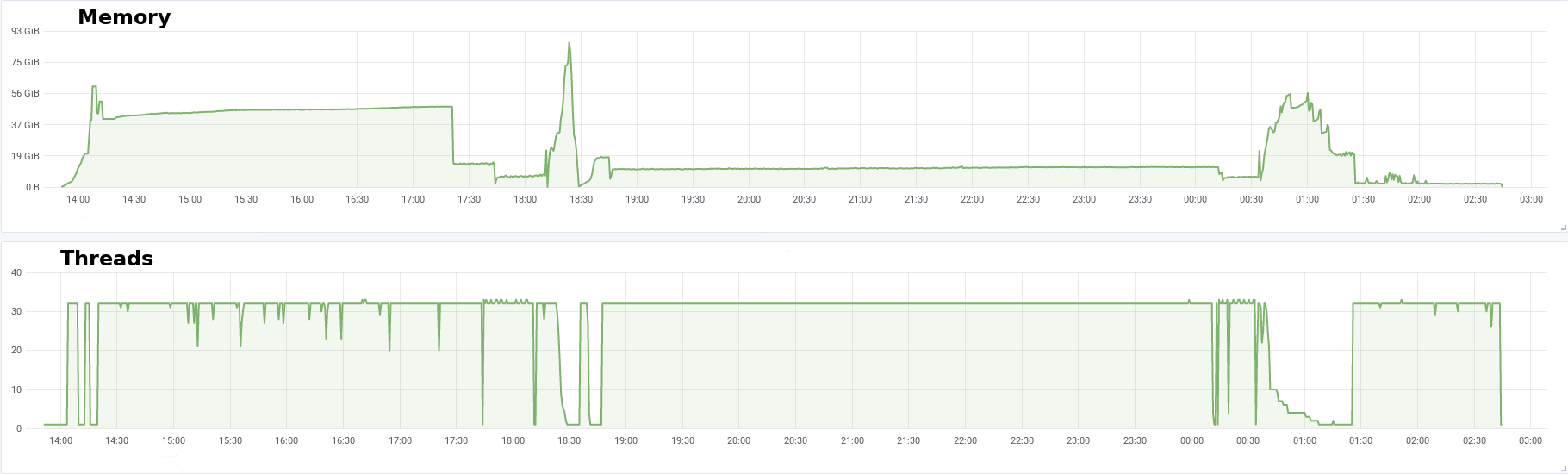
This job ran for ~13h and used up to 100GB of memory. Here is the profile of memory and running threads for this assembly:
The final result was an assembly of 137Mb with 357 contigs and a scaffold N50 of 6.34Mb.