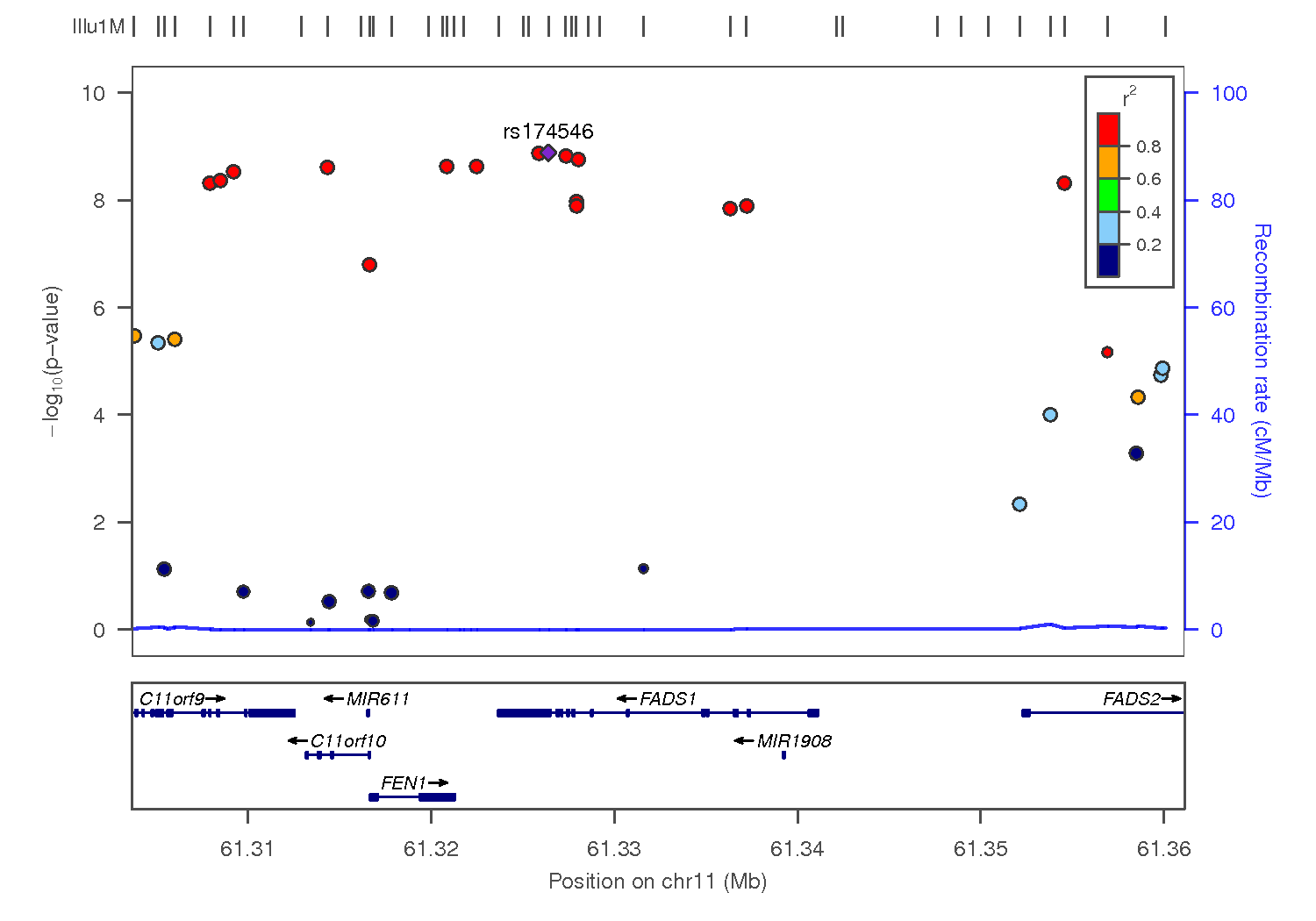
LocusZoom is a tool for visualizing results of genome wide association studies at an individual locus along with other relevant information like gene models, linkage disequillibrium coefficients, and estimated local recombination rates.
LocusZoom uses association results in METAL or EPACTS formatted files along with it's own source of supporting data (see below) to generate graphs.
$LOCUSZOOM_TEST_DATALocusZoom contains a number of data files used in generating graph annotations:
All data for LocusZoom is stored in /fdb/locuszoom/[version]. LocusZoom has been configured to automatically find all required information.
Allocate an interactive session and run the program. Sample session:
[user@biowulf]$ sinteractive
salloc.exe: Pending job allocation 46116226
salloc.exe: job 46116226 queued and waiting for resources
salloc.exe: job 46116226 has been allocated resources
salloc.exe: Granted job allocation 46116226
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn3144 are ready for job
[user@cn3144]$ module load locuszoom
[user@cn3144]$ locuszoom -h
locuszoom -h
+---------------------------------------------+
| LocusZoom 1.3 (06/20/2014) |
| Plot regional association results |
| from GWA scans or candidate gene studies |
+---------------------------------------------+
usage: locuszoom [options]
-h, --help
show this help message and exit
--metal <string>
Metal file.
[...snip...]
Draw a diagram of the associations between SNPs and HDL observed in Kathiresan et al, 2009 around the FADS1 gene
[user@cn3144]$ locuszoom \
--metal /usr/local/apps/locuszoom/TEST_DATA/examples/Kathiresan_2009_HDL.txt \
--refgene FADS1
Exit the interactive session
[user@cn3144]$ exit salloc.exe: Relinquishing job allocation 46116226 [user@biowulf]$
Create a batch input file (e.g. locuszoom.sh), which uses the input file 'locuszoom.in'. For example:
#! /bin/bash
#SBATCH --mail-type=END
# this is locuszoom.sh
set -e
function fail {
echo "$@" >&2
exit 1
}
module load locuszoom/1.3 || fail "Could not load locuszoom module"
mf=$LOCUSZOOM_TEST_DATA/examples/Kathiresan_2009_HDL.txt
locuszoom --metal=$mf --refgene FADS1 --pop EUR --build hg19 \
--source 1000G_March2012 \
--gwas-cat whole-cat_significant-only
Submit this job using the Slurm sbatch command.
sbatch [--cpus-per-task=#] [--mem=#] locuszoom.sh
Create a swarmfile (e.g. locuszoom.swarm). For example:
locuszoom --metal=$LOCUSZOOM_TEST_DATA/examples/Kathiresan_2009_HDL.txt \ --refgene FADS1 locuszoom --metal=$LOCUSZOOM_TEST_DATA/examples/Kathiresan_2009_HDL.txt \ --refgene PLTP locuszoom --metal=$LOCUSZOOM_TEST_DATA/examples/Kathiresan_2009_HDL.txt \ --refgene ANGPTL4
Submit this job using the swarm command.
swarm -f locuszoom.swarm [-g #] [-t #] --module locuszoomwhere
| -g # | Number of Gigabytes of memory required for each process (1 line in the swarm command file) |
| -t # | Number of threads/CPUs required for each process (1 line in the swarm command file). |
| --module locuszoom | Loads the locuszoom module for each subjob in the swarm |