MUSCLE is a popular multiple alignment program with good performance and accuracy. It can align hundreds of sequences quickly and has a simple command line interface with few options. ?Compared to previous versions, Muscle v5 is much more accurate, is often faster, and scales to much larger datasets. At the time of writing (late 2021), Muscle v5 has the highest scores on multiple alignment benchmarks including Balibase, Bralibase, Prefab and Balifam. It can align tens of thousands of sequences with high accuracy on a low-cost commodity computer (say, an 8-core Intel CPU with 32 Gb RAM). On large datasets, Muscle v5 is 20-30% more accurate than MAFFT and Clustal-Omega.
Allocate an interactive session and run the program. Sample session:
[user@biowulf]$ sinteractive --cpus-per-task=16 salloc.exe: Pending job allocation 46116226 salloc.exe: job 46116226 queued and waiting for resources salloc.exe: job 46116226 has been allocated resources salloc.exe: Granted job allocation 46116226 salloc.exe: Waiting for resource configuration salloc.exe: Nodes cn3144 are ready for job [user@cn3144 ~]$ module load muscle [+] Loading muscle 5.0.1428 [user@cn3144 ~]$ cp /usr/local/apps/muscle/sample_data/globins13.fa . [user@cn3144 ~]$ muscle -threads $SLURM_CPUS_PER_TASK -align globins25.fa -output globins25.muscle.out muscle 5.0.1428_linux64 396Gb RAM, 72 cores (C) Copyright 2004-2021 Robert C. Edgar. https://drive5.com 00:00 24Mb CPU has 72 cores, defaulting to 20 threads WARNING: Max OMP threads 16 02:00 1.6Gb 100.0% Calc posteriors 05:44 2.6Gb 100.0% Consistency (1/2) 09:30 2.6Gb 100.0% Consistency (2/2) 09:30 2.6Gb 100.0% UPGMA5 10:01 2.6Gb 100.0% Refining [user@cn3144 ~]$ exit salloc.exe: Relinquishing job allocation 46116226 [user@biowulf ~]$
Create a batch input file (e.g. Muscle.sh). For example:
#!/bin/bash set -e module load muscle cd /data/$USER/somedir muscle -threads $SLURM_CPUS_PER_TASK -align inputseqs.fa -output muscle_output
Submit this job using the Slurm sbatch command.
sbatch [--mem=#] --cpus-per-task=20 Muscle.sh
Create a swarmfile (e.g. Muscle.swarm). For example:
muscle -threads $SLURM_CPUS_PER_TASK -align file1.fa -output file1.out muscle -threads $SLURM_CPUS_PER_TASK -align file2.fa -output file2.out [....]
Submit this job using the swarm command.
swarm -f Muscle.swarm [-g #] -t 16 --module musclewhere
| -g # | Number of Gigabytes of memory required for each process (1 line in the swarm command file) |
| -t 16 | Number of CPUs to use for a single muscle command |
| --module Muscle | Loads the Muscle module for each subjob in the swarm |
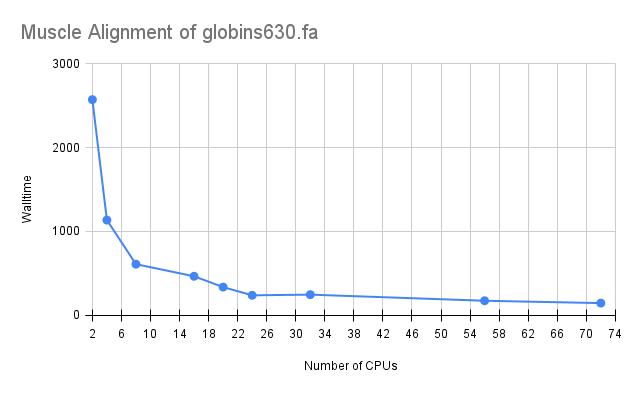
Benchmarks were run with input sequence file globins630.fa (available in /usr/local/apps/muscle/sample_data on Biowulf, and from the Hmmer tutorial) on Intel(R) Xeon(R) Gold 6140 CPU @ 2.30GHz ('x6140' nodes on Biowulf norm partition). This job requires about 2 GB memory.
Based on the benchmarks below, it is appropriate to submit to 20 or 24 CPUs for this muscle alignment. Increasing the allocated CPUs beyond 24 results in very little performance improvement, and is therefore inefficient.