PeakRanger is a multi-purporse software suite for analyzing next-generation sequencing (NGS) data. The suite contains the following tools:
| Tool | Description |
|---|---|
| nr | noise rate estimator. Estimates signal to noise ratio which is an indicator for ChIP enrichment |
| lc | library complexity calculator. Calculates the ratio of unique reads over total reads. Only accepts bam files. |
| wig | coverage file generator. Generates variable step format wiggle file |
| wigpe | coverage file generator. Generates bedGraph format wiggle file and supports spliced alignments and thus only supports bam files |
| ranger | ChIP-Seq peak caller. It is able to identify enriched genomic regions while at the same time discover summits within these regions. |
| ccat | ChIP-Seq peak caller. Tuned for the discovery of broad peaks |
| bcp | ChIP-Seq peak caller. Tuned for the discovery of broad peaks. |
$PEAKRANGER_TEST_DATAAllocate an interactive session and run the program. Sample session:
[user@biowulf]$ sinteractive --mem=8g
salloc.exe: Pending job allocation 46116226
salloc.exe: job 46116226 queued and waiting for resources
salloc.exe: job 46116226 has been allocated resources
salloc.exe: Granted job allocation 46116226
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn3144 are ready for job
[user@cn3144 ~]$ module load peakranger
[user@cn3144 ~]$ cp -L $PEAKRANGER_TEST_DATA/* .
[user@cn3144 ~]$ ls -lh
-rw-rw-r-- 1 user group 869M Mar 6 14:45 ENCFF374UMR.bam <-- CTCF (GrCh38)
-rw-rw-r-- 1 user group 793M Mar 6 14:46 ENCFF700UBO.bam <-- Control
[user@cn3144 ~]$ peakranger nr --data ENCFF374UMR.bam --control ENCFF700UBO.bam --format bam
...
Estimated noise rate:0.899125
[user@cn3144 ~]$ peakranger wig --format bam --data ENCFF374UMR.bam --output ENCFF374UMR.wig
[user@cn3144 ~]$ # ranger is for narrow peaks
[user@cn3144 ~]$ peakranger ranger --data ENCFF374UMR.bam --control ENCFF700UBO.bam --format bam \
--thread 2 --output ENCFF374UMR
Warning: chrEBV only contains reads in the positive strand. The chromosome is removed.
Warning: chrUn_KI270379v1 only contains reads in the negative strand. The chromosome is removed.
Warning: No reads were found in chrEBV of the treatment dataset. The chromosome is removed.
Warning: No reads were found in chrUn_KI270379v1 of the treatment dataset. The chromosome is removed.
[user@cn3144 ~]$ wc -l ENCFF374UMR_region.bed
41305 ENCFF374UMR_region.bed
[user@cn3144 ~]$ exit
salloc.exe: Relinquishing job allocation 46116226
[user@biowulf ~]$
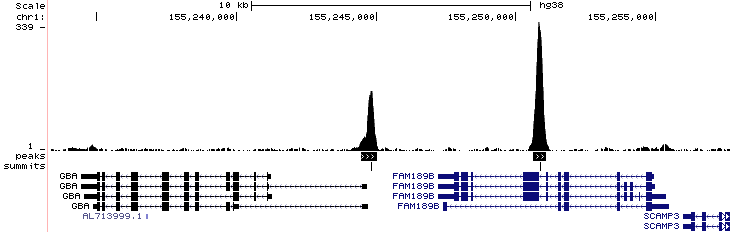
Here is an example of the resulting wig file after conversion to BigWig along with the peak and summit calls (chr1:155,233,328-155,257,703)
Note that the --report option will generate a separate plot for
each peak if R is on the path. All the plots will be in a single directory
along with a single html file linking all the images. This is
not recommended for a ChIP-Seq analysis such as this with
>40k peaks since it (a) will take 9h to finish compared to minutes when run
without generating reports and (b) will create a difficult to handle directory
with >40k files.
Create a batch input file (e.g. peakranger.sh) for processing each experiment/control pair. For example:
#!/bin/bash
expt="${1:-none}"
ctrl="${2:-none}"
[[ "${expt}" == "none" || "${ctrl}" == "none" ]] && exit 1
module load peakranger/1.18 || exit 1
module load ucsc samtools || exit 1
peakranger nr --data "${expt}" --control "${ctrl}" --format bam > "${expt%bam}.noiserate"
peakranger wig --format bam --data "${expt}" --output "${expt%bam}wig"
samtools view -H ENCFF374UMR.bam | grep '^@SQ' | tr ':' '\t' | cut -f3,5 > chroms
wigToBigWig -clip "${expt%bam}wig" chroms "${expt%bam}bw"
peakranger ranger --data "${expt}" --control "${ctrl}" --format bam \
--thread $SLURM_CPUS_PER_TASK --output ${expt%.bam}
Submit this job using the Slurm sbatch command.
sbatch --cpus-per-task=4 --mem=8g peakranger.sh ENCFF374UMR.bam ENCFF700UBO.bam
Create a swarmfile (e.g. peakranger.swarm). For example:
peakranger nr --data expt1.bam --control control.bam --format bam > expt1.noiserate peakranger nr --data expt2.bam --control control.bam --format bam > expt2.noiserate peakranger nr --data expt3.bam --control control.bam --format bam > expt3.noiserate
Submit this job using the swarm command.
swarm -f peakranger.swarm [-g #] [-t #] --module peakrangerwhere
| -g # | Number of Gigabytes of memory required for each process (1 line in the swarm command file) |
| -t # | Number of threads/CPUs required for each process (1 line in the swarm command file). |
| --module peakranger | Loads the peakranger module for each subjob in the swarm |