peddy is used to compare sex and familial relationships given in a PED file with those inferred from a VCF file. This is done by sampling 25000 sites plus chrX from the VCF file to estimate relatedness, heterozygosity, sex and ancestry. It uses data from the thousand genome project.
$PEDDY_TEST_DATAAllocate an interactive session and run the program. Sample session:
[user@biowulf]$ sinteractive --cpus-per-task=2 --gres=lscratch:10
salloc.exe: Pending job allocation 46116226
salloc.exe: job 46116226 queued and waiting for resources
salloc.exe: job 46116226 has been allocated resources
salloc.exe: Granted job allocation 46116226
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn3144 are ready for job
[user@cn3144]$ cd /lscratch/$SLURM_JOB_ID
[user@cn3144]$ module load peddy
[user@cn3144]$ cp -r $PEDDY_TEST_DATA/data .
[user@cn3144]$ peddy -p $SLURM_CPUS_PER_TASK --plot --prefix ceph-1463 \
data/ceph1463.peddy.vcf.gz data/ceph1463.ped
2018-05-21 08:14:22 cn4242 peddy.cli[35381] INFO Running Peddy version 0.4.8
2018-05-21 08:14:23 cn4242 peddy.cli[35381] INFO ped_check
2022-10-13 21:04:54 cn4291 peddy.peddy[83025] INFO plotting
2022-10-13 21:04:55 cn4291 peddy.cli[83025] INFO ran in 2.0 seconds
2022-10-13 21:04:55 cn4291 peddy.cli[83025] INFO het_check
2022-10-13 21:04:56 cn4291 peddy.pca[83025] INFO loaded and subsetted thousand-genomes genotypes (shape: (2504, 2724)) in 0.3 seconds
2022-10-13 21:04:56 cn4291 peddy.pca[83025] INFO ran randomized PCA on thousand-genomes samples at 2724 sites in 0.5 seconds
2022-10-13 21:04:57 cn4291 peddy.pca[83025] INFO Projected thousand-genomes genotypes and sample genotypes and predicted ancestry via SVM in 0.1 seconds
2022-10-13 21:04:57 cn4291 peddy.cli[83025] INFO ran in 2.7 seconds
2022-10-13 21:04:57 cn4291 peddy.cli[83025] INFO sex_check
2022-10-13 21:04:57 cn4291 peddy.peddy[83025] INFO sex-check: 0 skipped / 814 kept
2022-10-13 21:04:57 cn4291 peddy.cli[83025] INFO ran in 0.1 seconds
[user@cn3144]$ ls -lh
-rw-r--r-- 1 user group 169K May 21 08:14 ceph-1463.background_pca.json
-rw-r--r-- 1 user group 2.0K May 21 08:14 ceph-1463.het_check.csv
-rw-r--r-- 1 user group 18K May 21 08:14 ceph-1463.het_check.png
-rw-r--r-- 1 user group 211K May 21 08:14 ceph-1463.html
-rw-r--r-- 1 user group 118K May 21 08:14 ceph-1463.pca_check.png
-rw-r--r-- 1 user group 13K May 21 08:14 ceph-1463.ped_check.csv
-rw-r--r-- 1 user group 108K May 21 08:14 ceph-1463.ped_check.png
-rw-r--r-- 1 user group 96 May 21 08:14 ceph-1463.ped_check.rel-difference.csv
-rw-r--r-- 1 user group 1.7K May 21 08:14 ceph-1463.peddy.ped
-rw-r--r-- 1 user group 835 May 21 08:14 ceph-1463.sex_check.csv
-rw-r--r-- 1 user group 25K May 21 08:14 ceph-1463.sex_check.png
[user@cn3144]$
[user@cn3144]$ exit
salloc.exe: Relinquishing job allocation 46116226
[user@biowulf]$
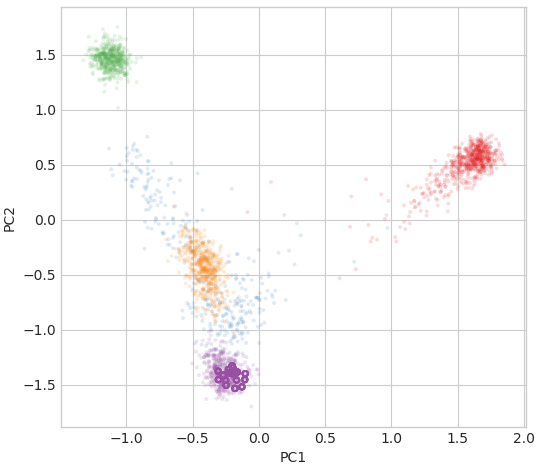
peddy creates several plots, data tables, and a sumamry report in html. This includes, for example, the following check on ancestry showing reported ancestry in the pedigree overlayed on a PCA of background genomes.
Create a batch input file (e.g. peddy.sh), which uses the data in $PEDDY_TEST_DATA.
For example:
#! /bin/bash
# this file is peddy.batch
module load peddy/0.4.8 || exit 1
td=/usr/local/apps/peddy/TEST_DATA/data
cp -r $PEDDY_TEST_DATA/data .
peddy -p $SLURM_CPUS_PER_TASK --plot --prefix ceph-1463 \
data/ceph1463.peddy.vcf.gz data/ceph1463.ped
Submit this job using the Slurm sbatch command.
sbatch --cpus-per-task=4 --mem=4g peddy.sh
Create a swarmfile (e.g. peddy.swarm). For example:
peddy -p $SLURM_CPUS_PER_TASK --plot --prefix fam1 fam1/fam1.vcf.gz fam1/fam1.ped peddy -p $SLURM_CPUS_PER_TASK --plot --prefix fam2 fam2/fam2.vcf.gz fam2/fam2.ped peddy -p $SLURM_CPUS_PER_TASK --plot --prefix fam3 fam3/fam3.vcf.gz fam3/fam3.ped
Submit this job using the swarm command.
swarm -f peddy.swarm -g 4 -t 4 --module peddy/0.4.8where
| -g # | Number of Gigabytes of memory required for each process (1 line in the swarm command file) |
| -t # | Number of threads/CPUs required for each process (1 line in the swarm command file). |
| --module peddy | Loads the peddy module for each subjob in the swarm |