Poretools is a toolkit for manipulating and exploring nanopore sequencing data sets. Poretools operates on individual FAST5 files, directory of FAST5 files, and tar archives of FAST5 files.
$PORETOOLS_TEST_DATAAllocate an interactive session and run the program. Sample session:
[user@biowulf]$ sinteractive --mem=6g
salloc.exe: Pending job allocation 46116226
salloc.exe: job 46116226 queued and waiting for resources
salloc.exe: job 46116226 has been allocated resources
salloc.exe: Granted job allocation 46116226
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn3144 are ready for job
[user@cn3144]$ module load poretools
[user@cn3144]$ poretools --help
usage: poretools [-h] [-v]
{combine,fastq,fasta,...,yield_plot,occupancy,organise}
...
optional arguments:
-h, --help show this help message and exit
-v, --version Installed poretools version
[sub-commands]:
{combine,fastq,fasta,...,yield_plot,occupancy,organise}
combine Combine a set of FAST5 files in a TAR achive
fastq Extract FASTQ sequences from a set of FAST5 files
fasta Extract FASTA sequences from a set of FAST5 files
stats Get read size stats for a set of FAST5 files
hist Plot read size histogram for a set of FAST5 files
events Extract each nanopore event for each read.
readstats Extract signal information for each read over time.
tabular Extract the lengths and name/seq/quals from a set of
FAST5 files in TAB delimited format
nucdist Get the nucl. composition of a set of FAST5 files
metadata Return run metadata such as ASIC ID and temperature
from a set of FAST5 files
index Tabulate all file location info and metadata such as
ASIC ID and temperature from a set of FAST5 files
qualdist Get the qual score composition of a set of FAST5 files
qualpos Get the qual score distribution over positions in
reads
winner Get the longest read from a set of FAST5 files
squiggle Plot the observed signals for FAST5 reads.
times Return the start times from a set of FAST5 files in
tabular format
yield_plot Plot the yield over time for a set of FAST5 files
occupancy Inspect pore activity over time for a set of FAST5
files
organise Move FAST5 files into a useful folder hierarchy
[user@cn3144]$ cp ${PORETOOLS_TEST_DATA}/ERA484348_014370_subset.tar .
[user@cn3144]$ ls -lh ERA484348_014370_subset.tar
-rw-r--r-- 1 user group 2.1G Jun 30 08:42 ERA484348_014370_subset.tar
Extract fastq format sequences from all the pass reads in the example data set. Note that poretools can work on a tar archive directly - no need to extract the archive and create large numbers of small files that can degrade file system performance.
[user@cn3144]$ poretools fastq ERA484348_014370_subset.tar | gzip -c - > 014370.fastq.gz [user@cn3144]$ ls -lh 014370.fastq.gz -rw-r--r-- 1 user group 13M Jun 30 08:51 014370.fastq.gz
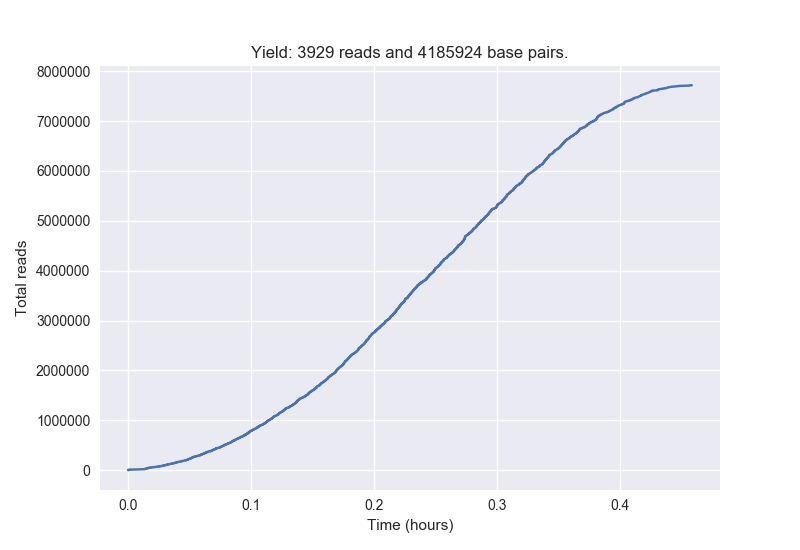
Create a collector's curve of yield
[user@cn3144]$ poretools yield_plot --plot-type reads --saveas yield.png \
ERA484348_014370_subset.tar
Read size information
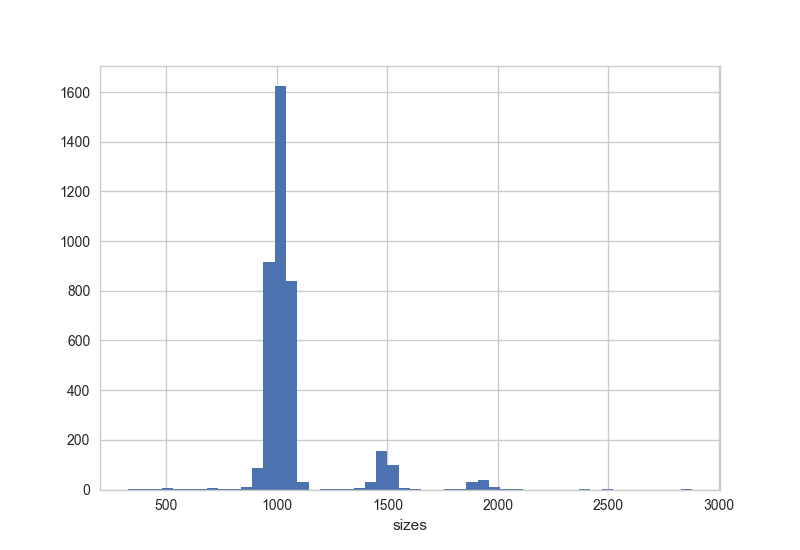
[user@cn3144]$ poretools stats ERA484348_014370_subset.tar total reads 11787 total base pairs 12470421 mean 1057.98 median 1013 min 320 max 2877 N25 1060 N50 1019 N75 981 [user@cn3144]$ poretools stats --type fwd ERA484348_014370_subset.tar total reads 3929 total base pairs 4112608 mean 1046.73 median 1001 min 320 max 2833 N25 1044 N50 1007 N75 973 [user@cn3144]$ poretools hist --saveas size.png --theme-bw ERA484348_014370_subset.tar
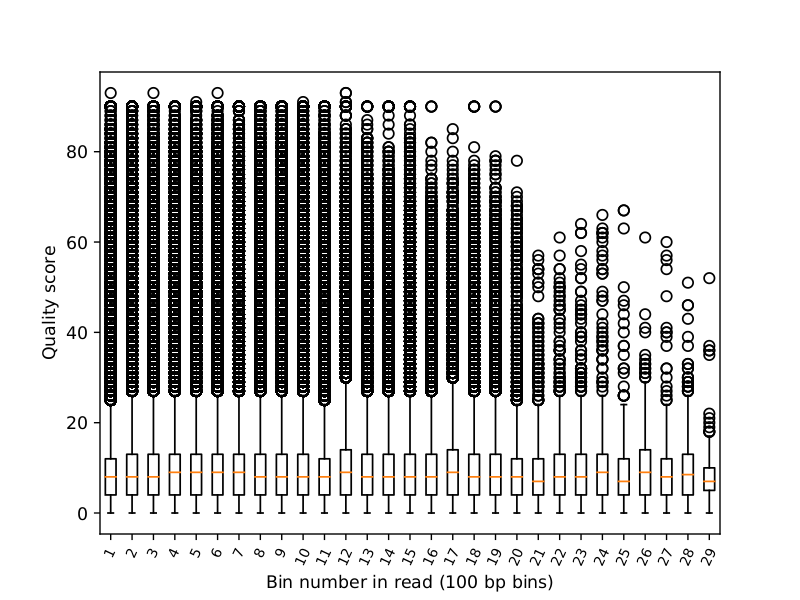
Quality score distribution by position
[user@cn3144]$ poretools qualpos --saveas qual.pdf \ --bin-width 100 ERA484348_014370_subset.tar
Exit the interactive session
[user@cn3144]$ exit salloc.exe: Relinquishing job allocation 46116226 [user@biowulf]$
Create a batch input file (e.g. poretools.sh), which uses the input file 'poretools.in'. For example:
#! /bin/bash # this file is poretools.batch module load poretools/0.6.1a1 || exit 1 poretools stats ERA484348_014370_subset.tar > stats poretools qualpos --saveas qual.png --bin-width 100 ERA484348_014370_subset.tar
Submit this job using the Slurm sbatch command.
sbatch --time=20 --cpus-per-task=2 --mem=6g poretools.sh
Create a swarmfile (e.g. poretools.swarm). For example:
poretools qualpos --saveas qual_s1.png --bin-width 100 s1.tar poretools qualpos --saveas qual_s2.png --bin-width 100 s2.tar poretools qualpos --saveas qual_s3.png --bin-width 100 s3.tar
Submit this job using the swarm command.
swarm -f poretools.swarm [-g #] [-t #] --module poretoolswhere
| -g # | Number of Gigabytes of memory required for each process (1 line in the swarm command file) |
| -t # | Number of threads/CPUs required for each process (1 line in the swarm command file). |
| --module poretools | Loads the poretools module for each subjob in the swarm |