From the scallop repository:
Scallop is an accurate reference-based transcript assembler. Scallop features its high accuracy in assembling multi-exon transcripts as well as lowly expressed transcripts. Scallop achieves this improvement through a novel algorithm that can be proved preserving all phasing paths from paired-end reads, while also achieves both transcripts parsimony and coverage deviation minimization.
$SCALLOP_TEST_DATAAllocate an interactive session and run the program. Sample session:
[user@biowulf]$ sinteractive --gres=lscratch:10 --mem=7g
salloc.exe: Pending job allocation 46116226
salloc.exe: job 46116226 queued and waiting for resources
salloc.exe: job 46116226 has been allocated resources
salloc.exe: Granted job allocation 46116226
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn3144 are ready for job
[user@cn3144]$ module load scallop
[user@cn3144]$ cd /lscratch/$SLURM_JOB_ID
[user@cn3144]$ # copy some example data - paired end RNA-Seq (101nt) of human
# skin aligned with STAR for about a quarter of chr8 including Myc.
# this is stranded RNA-Seq data
[user@cn3144]$ cp $SCALLOP_TEST_DATA/ENCSR862RGX.bam .
[user@cn3144]$ ls -lh ENCSR862RGX.bam
-rw-r--r-- 1 user group 81M Apr 18 07:30 ENCSR862RGX.bam
[user@cn3144]$ # run scallop
[user@cn3144]$ scallop --library_type second --min_transcript_length 300 \
-i ENCSR862RGX.bam -o ENCSR862RGX.gtf
command line: scallop --library_type second --min_transcript_length 300 -i ENCSR862RGX.bam -o ENCSR862RG X.gtf
Bundle 0: tid = 7, #hits = 93, #partial-exons = 22, range = chr8:101915822-102124299, orient = + (93, 0, 0)
process splice graph gene.0.0 type = 1, vertices = 3, edges = 0
process splice graph gene.0.1 type = 0, vertices = 5, edges = 4
process splice graph gene.0.2 type = 1, vertices = 3, edges = 0
process splice graph gene.0.3 type = 1, vertices = 3, edges = 0
[...snip...]
[user@cn3144]$ wc -l ENCSR862RGX.gtf
5057 ENCSR862RGX.gtf
[user@cn3144]$ egrep '"gene.1.5"' ENCSR862RGX.gtf | head -3
chr8 scallop transcript 102204502 102239040 1000 + . gene_id "gene.1.5"; transcript_id "gene.1.5.2"; RPKM "22.4072"; cov "1.9556";
chr8 scallop exon 102204502 102205959 1000 + . gene_id "gene.1.5"; transcript_id "gene.1.5.2"; exon "1";
chr8 scallop exon 102208095 102208285 1000 + . gene_id "gene.1.5"; transcript_id "gene.1.5.2"; exon "2";
[user@cn3144]$ exit
salloc.exe: Relinquishing job allocation 46116226
[user@biowulf]$
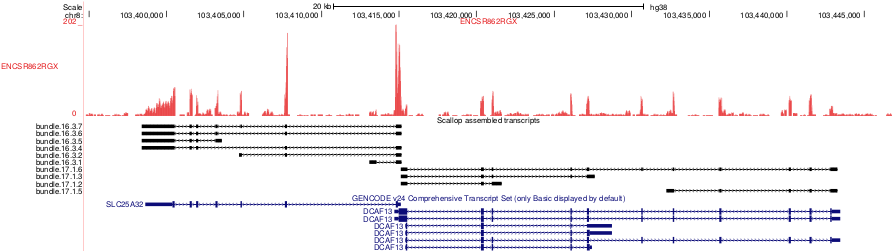
Example of scallop assembled transcripts for chr8:103,394,722-103,446,765. The scallop transcripts are shown in black. Gencode v24 annotation is shown in blue.
Create a batch input file (e.g. scallop.sh), which uses the input file 'scallop.in'. For example:
#! /bin/bash
module load scallop/0.10.2 || exit 1
scallop --verbose 0 --library_type second \
-i ENCSR862RGX.bam -o ENCSR862RGX.gtf
Submit this job using the Slurm sbatch command.
sbatch --cpus-per-task=2 --mem=7g scallop.sh
Create a swarmfile (e.g. scallop.swarm). For example:
scallop --verbose 0 --library_type second -i sample1.bam -o sample1.gtf scallop --verbose 0 --library_type second -i sample2.bam -o sample2.gtf scallop --verbose 0 --library_type second -i sample3.bam -o sample3.gtf
Submit this job using the swarm command.
swarm -f scallop.swarm -g 7 -t 2 --module scallop/0.10.2where
| -g # | Number of Gigabytes of memory required for each process (1 line in the swarm command file) |
| -t # | Number of threads/CPUs required for each process (1 line in the swarm command file). |
| --module scallop | Loads the scallop module for each subjob in the swarm |