scVelo is a method to describe the rate of gene expression change for an individual gene at a given time point based on the ratio of its spliced and unspliced messenger RNA (mRNA). It avoids errors in the velocity estimates by solving the full transcriptional dynamics of splicing kinetics using a likelihood-based dynamical model. This generalizes RNA velocity to systems with transient cell states, which are common in development and in response to perturbations.
Allocate an interactive session and run the program. Sample session:
[user@biowulf]$ sinteractive --mem=4g
[user@cn0911 ~]$module load scvelo
[+] Loading scvelo 0.2.4 on cn0911
[+] Loading singularity 3.8.5 on cn0911
[user@cn0911 ~]$python-scvelo $SCVELO_SRC/tests/test_basic.py
[user@cn0911 ~]$python-scvelo
Python 3.8.12 (default, Oct 12 2021, 13:49:34)
[GCC 7.5.0] :: Anaconda, Inc. on linux
Type "help", "copyright", "credits" or "license" for more information.
>>> import numpy as np
>>> import matplotlib.pyplot as pl
>>> import scvelo as scv
>>> scv.settings.set_figure_params('scvelo', dpi_save=200, dpi=80, transparent=True)
>>> scv.settings.plot_prefix = 'scvelo_fig2_'
>>> scv.settings.verbosity = 2
>>> adata = scv.datasets.dentategyrus()
100%|██████████████████████████████████████████████████████████████████████████████████████| 23.7M/23.7M [00:00<00:00, 71.1MB/s]
>>> scv.pp.filter_and_normalize(adata, min_shared_cells=20, n_top_genes=2000)
Filtered out 11835 genes that are detected in less than 20 cells (shared).
Normalized count data: X, spliced, unspliced.
Extracted 2000 highly variable genes.
Logarithmized X
>>> scv.pp.moments(adata, n_neighbors=30, n_pcs=30)
computing neighbors
finished (0:00:13)
computing moments based on connectivities
finished (0:00:00)
>>> scv.tl.velocity(adata, vkey='steady_state_velocity', mode='steady_state')
computing velocities
finished (0:00:00)
>>> scv.tl.velocity_graph(adata, vkey='steady_state_velocity')
computing velocity graph (using 1/56 cores)
finished (0:00:04)
>>> scv.tl.recover_dynamics(adata)
recovering dynamics (using 1/56 cores)
finished (0:06:25)
>>> scv.tl.velocity(adata, mode='dynamical', vkey='dynamical_velocity')
computing velocities
finished (0:00:02)
>>> scv.tl.velocity_graph(adata, vkey='dynamical_velocity', variance_stabilization=True)
computing velocity graph (using 1/56 cores)
finished (0:00:08)
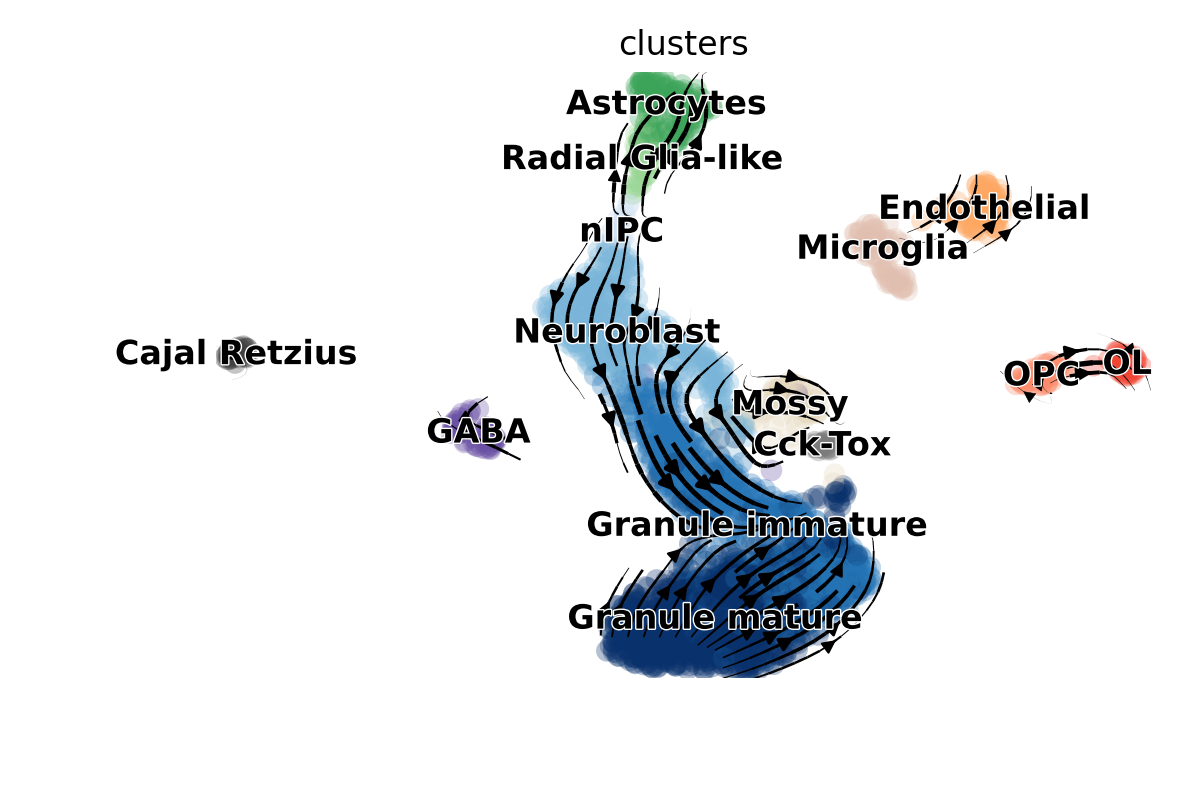
>>> scv.pl.velocity_embedding_stream(adata, vkey='dynamical_velocity')
computing velocity embedding
finished (0:00:08)
[user@cn0911 ~]$ exit salloc.exe: Relinquishing job allocation 46116226 [user@biowulf ~]$